
Cell-free DNA as a biomarker in cancer

 ,
, Abstract
Translational research of liquid biopsy is just at the edge of routine clinical application: an emerging validity of circulating tumor DNA (ctDNA) tests suggests its use for earlier cancer detection and better monitoring of minimal residual disease (MRD) and resistance development, thus offering earlier guidance for therapy choices with the intent to cure cancer. In this review, we focus on ctDNA as an advanced and standardized validated marker in liquid biopsy. We also discuss what will be needed to reach the new milestone of personalized (precision) medicine to be used as a common standard of care. We summarize recent developments of cell-free DNA (cfDNA) and its clinical use as a biomarker in cancer.
Keywords
INTRODUCTION
Key elements of clinical routine with tumor patients include cancer detection and diagnosis as well as monitoring tumor development and treatment response. Usually, an interdisciplinary approach is necessary with complementing areas, such as medical imaging, tissue biopsy, histopathology, surgery, chemotherapy, and radiotherapy. Over decades, measurements of protein tumor markers in the blood evolved to the current standard, but translational research offers improvement: circulating cell-free DNA (cfDNA) allows easy and early access to information on tumor evolution and treatment response at low risk of injury, infection, or wound healing, compared to repeated tissue biopsies or delayed medical imaging, thus enabling improved therapy decisions and avoidance of over-treatment which improves patients’ quality of life [Figures 1 and 2]. Circulating tumor cells (CTCs) were first described in an autopsy over 150 years ago by Ashworth, who found tumor cells in the blood microscopically identical to the tumor cells in metastatic skin lesions [Table 1][3]. When in 1889, over 130 years ago, the surgeon Stephen Paget presented his “seed and soil” theory, it was already known that tumor cells were able to disseminate via blood circulation. Metastatic cancer cells (“seeds”) can leave their site of origin and enter the bloodstream to grow as secondary tumors in favorable environments (“soil”) quite distant from the primary tumor[4]. With his model, Paget explained a 15-fold higher frequency of breast cancer metastasis to the liver compared to the spleen. Although only a very small fraction of tumor cells within the blood gives rise to secondary tumors, it is well accepted nowadays that circulating tumor cells (CTCs) indicate tumor progression and an increased risk of metastasis [Figure 1][6]. Over the last two decades, the detection and analysis of these CTCs have improved considerably enough to be tested in clinical settings [Table 1][28]. In 2004, Allard and colleagues were able to detect and count CTCs in blood from many patients with prostate, breast, ovarian, colorectal, lung, and other cancers[11]. In the same year, Cristofanilli and colleagues showed that an elevated number of CTCs before treatment of metastatic breast cancer can serve as an independent prognostic marker for a worse outcome, namely a shortened duration of both progression-free survival (PFS) and overall survival (OS)[12]. Molecular profiling of DNA extracted from CTCs from non-small cell lung cancer (NSCLC) patients during treatment allowed monitoring of tumor evolution by mutations in the epidermal growth factor receptor (EGFR) gene related or unrelated to therapy resistance[14]. As an alternative to the technical obstacles and the low sensitivity of fishing for the extremely rare CTCs, the cell-free fraction of blood can also be used to extract DNA, which then also allows the finding of tumor-specific mutations. In 1948, French scientists reported free nucleic acids in blood, but they did not relate them to any disease[5]. In 1977, Leon and colleagues used polyclonal antiserum from patients with systemic lupus erythematosus (SLE), an autoimmune disease generating antibodies against DNA, to detect increased values of free DNA in about half of their cancer patients[7]. Normal values for the other half of the patients were explained by the selection of patients before radiotherapy, but who already had their tumors surgically removed and/or had chemotherapy, resulting in an already reduced tumor mass. Despite significantly higher levels in patients with metastatic disease, no correlation was found regarding the size or location of the tumors. However, radiation therapy was able to decrease the levels of cfDNA significantly for lymphoma, lung, ovary, uterus, and cervical tumors, as well as to a lesser extent, glioma, breast, colon, and rectal tumors. Decreases in cfDNA levels went along with improved clinical conditions, such as lower tumor size and less pain, whereas increasing or unchanged levels correlated with worse conditions and a lack of response to treatment. The amount of ctDNA has been shown to markedly decrease after surgery or chemotherapy, suggesting the usefulness of ctDNA as a tumor marker [Figure 2 and Table 2][15]. The term “liquid biopsy” was originally coined in 2010 by Pantel and Alix-Panabières for several methods of CTC detection and analysis, but it was also used for ctDNA or any other analysis of tumor-derived material from biofluids, e.g. blood, cerebrospinal fluid (CSF), or urine[18,30]. Recently, these methods of tracking cancer in liquid biopsy are considered one of the milestones in cancer research of the last two decades[28].
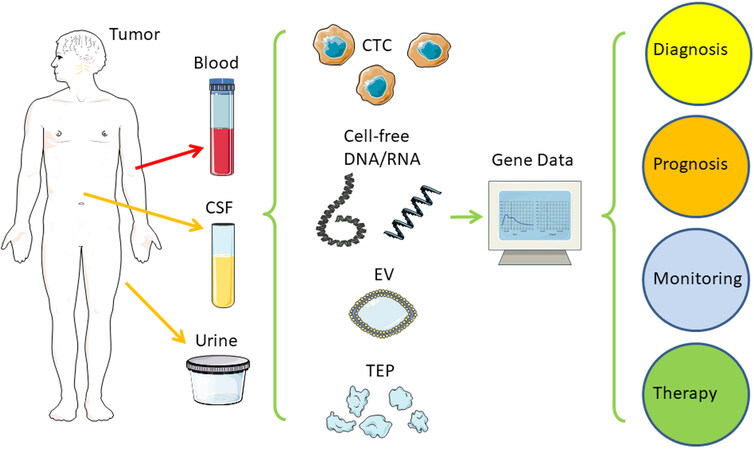
Figure 1. Liquid biopsy. Distant from the original tumor, samples from blood, CSF, or urine can serve as an easily acquired and low-risk source of tumor-derived nucleic acids (RNA and DNA) for further analysis to predict and monitor tumor progression and treatment response. CSF: Cerebrospinal fluid; CTC: circulating tumor cell; EV: extracellular vesicle; TEP: tumor educated platelets. Created/modified with SMART[1], licensed under Creative Commons Attribution 3.0 Unported License[2].
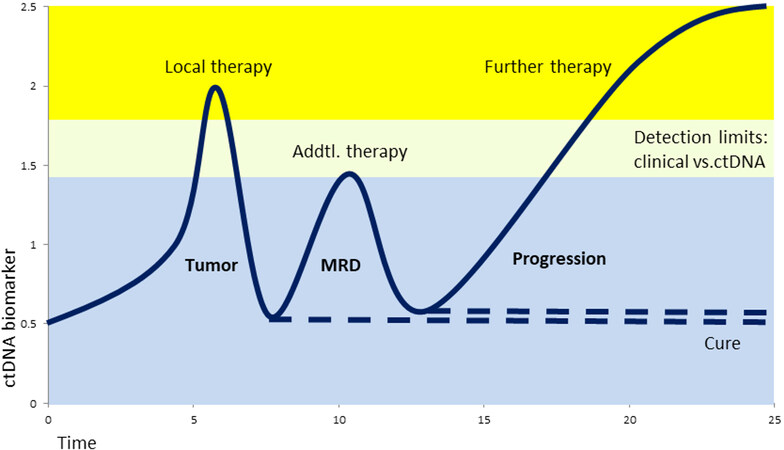
Figure 2. Hypothetical ctDNA biomarker levels during tumor development and therapy. After tumor removal, the biomarker level drops significantly and will remain low in the case the patient is cured. Early detection of minimal residual disease (MRD) by ctDNA allows additional treatment (e.g., chemo-, radio-, or hormone therapy) with the intent of cure, much earlier than with clinical imaging methods.
Historical timeframe and major developments of liquid biopsy
| Year | Author | Probe | Method | Tumor | Milestone |
| 1868 | Ashworth[3] | CTC | Microscopy, case report | Skin metastasis of unknown primary, “liquid autopsy” | First report on tumor cells in blood; post mortem; microscopically identical cells in metastatic lesions |
| 1889 | Paget[4] | CTC | Autopsy | Breast cancer, postulated | “Seed and soil” theory of cancer metastasis |
| 1948 | Mandel and Métais[5] | cfDNA | Blood analysis | Not related to cancer, healthy blood donors | First report of (cell-free) nucleic acids in blood |
| 1975 | Fidler[6] | CTC | Experimental metastasis assay | B16 melanoma cell lines | Only a small fraction of intravenously injected tumor cells give rise to metastasis in mouse models |
| 1977 | Leon et al.[7] | cfDNA | Radioimmunoassay for free DNA in serum | Various cancers | First report on increased cfDNA levels in some cancer patients; correlation with therapy response |
| 1989 | Lo et al.[8] | cfDNA | PCR of Y chromosome-specific sequence; cfDNA/blood | Not related to cancer | Sex determination of fetus in pregnant women |
| 2001 | Reya et al.[9] | CTC | Applying hematopoietic stem cell knowledge to heterogeneity of cancer cells | Solid tumors and leukemia, migratory CSC | Cancer stem cell theory |
| 2003 | Balaña et al.[10] | ctDNA | Methylation-specific PCR of MGMT, p16, DAPK, RASSF1A | GBM | Detection of methylated MGMT in serum highly predictive for response to BCNU chemotherapy |
| 2004 | Allard et al.[11] | CTC | CellSearch™ | Prostate, breast, ovarian, CCR, lung, and other cancers | Detection of CTCs in 7.5 mL of blood samples |
| 2004 | Cristofanilli et al.[12] | CTC | CellSearch™ Amount of CTC | Metastatic breast cancer | Independent predictive marker: reduced PFS and reduced OS |
| 2005 | Diehl et al.[13] | ctDNA | dPCR, BEAMing | Advanced CRC | APC mutations in plasma |
| 2008 | Maheswaran et al.[14] | CTC | Molecular profiling EGFR mutations | NSCL | Monitoring therapy |
| 2008 | Diehl et al. [15] | ctDNA | Mutations | Colorectal cancer | Amount and presence/absence as an independent tumor marker, monitoring |
| 2008 | Cohen et al.[16] | CTC | CellSearch Clinical study | Colorectal cancer | Enumerating CTC |
| 2008 | De Bono et al.[17] | CTC | Clinical study | Prostate Cancer | Enumerating, the first demonstration that CTCs are the most accurate and independent predictor of OS in metastatic prostate cancer |
| 2010 | Pantel and Alix-Panabières[18] | CTC | Concept of analyzing tumor cells in body fluids | All cancers | Coined the term “liquid biopsy” |
| 2010 | Calverly et al.[19] | TEP | Platelet mRNA | Metastatic NSCL | Downregulation of gene expression in platelets |
| 2013 | Dawson et al.[20] | CTC + ctDNA (comparison) | Side-by-side disease monitoring | Metastatic breast cancer undergoing treatment | Sensitivity: ctDNA>CTC ctDNA level correlates with treatment response |
| 2014 | Bettegowda et al.[21] | ctDNA | Digital PCR, sequencing | 14 tumor types | ctDNA detectable for most tumors outside the brain |
| 2014 | Sullivan et al.[22] | CTC | “Negative depletion” CTC-iChip (removing leukocytes from blood) | GBM (usually not metastatic) | Surprising and frequent detection of CTCs in brain tumors |
| 2015 | Mazel et al.[23] | CTC | CellSearch | Breast cancer | PD-L1 detection for immune checkpoint inhibition |
| 2016 | Tie et al.[24] | ctDNA | Sequencing | Colon cancer stage II | Detection of MRD after surgery; prediction of recurrence after chemotherapy |
| 2016 | Donaldson and Park[25] | ctDNA | Clinical studies | NSCLC | First FDA[29] and EMA approval to use ctDNA for EGFR-targeted therapy |
| 2018 | Cohen et al.[26] | ctDNA, plus proteins from blood | CancerSEEK, detecting mutations in 1933 loci of 16 genes; combined with protein tumor markers | 8 cancer types | Blood screening test for several common cancers |
| 2020 | Lennon et al.[27] | ctDNA, protein markers plus PET-CT | Prospective 16 gene locations, 8 tumor proteins, PET-CT | Multi-cancer screening of 10,000 women with no known cancer | Multi-cancer blood testing combined with PET-CT |
Principles of liquid biopsy (ctDNA or CTC) in clinical settings
| Liquid Biopsy | Pro | Con | Clinical utility |
| CTC | Sufficient sensitivity for some advanced-stage cancers, incl. metastases | Sensitivity limited: - Early-stage cancers - Cancer screening - Many advanced-stage cancers | Prognosis in many metastatic settings: - Breast - Prostate - Colorectal carcinoma |
| Validated for specific applications (enumeration) | Not fully standardized, difficult methods / for many applications; rare CTCs; very few, centralized high-tech laboratories needed | Prediction of relapse after treatment | |
| High specificity (mutations) | Sophisticated technology, no easy and common standards | Live CTCs useful for drug screening and functional assays | |
| ctDNA | Standardized, common methods: - blood drawing/handling (routine in hospitals AND practice) - DNA extraction (decentralized laboratories) PCR/sequencing/analysis | Sensitivity too low: - Early-stage cancers - Cancer screening - Some advanced-stage cancers | - Diagnosis - Prognosis - Monitoring: Tumor evolution Relapse Therapy resistance (e.g., EGFR mutations) (all complementing tissue biopsy, medical imaging) |
| High specificity (often close to 100% with tumor-related mutations) | Limited biological and clinical relevance of many detected mutations (not every mutation is relevant or druggable) | Personalized, precision medicine: early detection of druggable mutations | |
| Fast/real-time monitoring of relapse or resistance after therapy | |||
| Medium sensitivity for advanced cancer |
Over the last two decades, liquid biopsy has included numerous methods to search for cancer-derived markers by analyzing biofluids, e.g. blood, CSF, or urine, i.e. samples taken quite distant from both the original tumor and potential metastases, generally with much easier and less risky access [Figure 1][31]. This led to a new definition of liquid biopsy with different circulating biomarkers in different body fluids[30]. Basically, liquid biopsy targets circulating tumor cells (CTC), or circulating tumor DNA (ctDNA), which is part of the whole cfDNA [Table 2]. Other examples include the analysis of extracellular vesicles (EVs) and RNA, such as microRNA (miRNA, miR) or, very recently, circular RNA (circRNA)[32]. The common goals are the use in screening, i.e. early detection and diagnosis of cancer with the chance for earlier and more curative treatments, as well as the use in monitoring after therapy and a better quality of life. The vision is a quick, reliable, and easy-to-repeat test for all patients to track tumor progression in real-time, as well as monitor the response to treatment to allow earlier detection of resistance, relapse, and metastasis and adapt clinical decisions accordingly. In addition, the presence of tumor markers after initially successful therapy should confirm minimal residual disease (MRD) in the absence of other markers and lead to additional treatments before the tumor becomes incurable[33]. The current standard for most solid cancers consists of a more invasive tissue biopsy and/or surgical removal of all or most parts of the tumor with a thorough microscopic analysis by a tumor pathologist. Genetic molecular profiling of the primary tumor tissue may be included. In 2016, a new WHO classification of tumors of the nervous system introduced molecular and genetic profiling as standard practice for the analysis of some brain tumors such as medulloblastomas and glioblastomas[34]. Depending on the tumor location and the multi-morbidity of a patient, obtaining tumor tissue repeatedly can be difficult and unsuitable for long-term monitoring of tumor development. Furthermore, transthoracic lung biopsies can lead to, although rare, iatrogenic dissemination of cancer cells[35], and fine-needle biopsies have been reported to increase CTCs in prostate cancer[36]. The snapshot of a tissue biopsy from a primary tumor may also miss minor, but progressing, tumor clones; it may also not represent metastatic clones with perhaps different molecular profiles for treatment resistances[37]. Therefore, liquid biopsy carries the potential to be the next gold standard for monitoring cancer progression. Following various research approaches to the detection and cultivation of CTCs, also leading to a better understanding of tumor progression and metastasis, hundreds of emerging clinical studies are continuing to develop new standards for monitoring melanoma, breast, prostate, colon, and head and neck cancers[38-46]. In this review, we focus on another part of liquid biopsy, cfDNA, which is reliably much easier to access and does not need elaborate methodologies to capture the extremely rare CTCs.
LIQUID BIOPSY MARKERS
ctDNA and CTCs are currently more established than EVs and tumor-educated platelets (TEP) as tools in liquid biopsy [Figures 1 and 2 and Table 1]. In this review, our main focus is on ctDNA and its clinical application and potential. For completion and comparison, we mention CTCs, EVs, and TEPs only briefly.
Cell-free DNA and circulating tumor DNA
The total amount of non-cellular DNA in samples such as blood is called cfDNA, which for tumor patients contains all tumor-derived DNA in various amounts, i.e. ctDNA.
cfDNA in blood is highly fragmented with a peak of 166 bp length and many smaller peaks of 10 bp less or more in healthy individuals. Interestingly, the fragment size of the tumor-derived ctDNA appears to be shifted to a lower peak at 146 bp, which allows for selection strategies to increase the relative amount of tumor-derived ctDNA and to increase the sensitivity. Most of the cfDNA is considered to come from leukocytes, such as granulocytes, lymphocytes, and monocytes, and to a lesser extend from other cells (muscle cells, endothelial cells, epithelial cells, fibroblasts, mesenchymal stem cells, and hepatocytes)[47]; interestingly, physical activity can trigger cfDNA release almost exclusively from granulocytes (up to 2-20-fold increase) and may affect liquid biopsy[47]. Tumor-derived ctDNA can come from apoptotic or necrotic parts of the primary tumor, as well as from metastases and CTCs. ctDNA often represents only an extremely small fraction of the total cfDNA, especially at early stages and after successful therapies. Therefore, detection of low concentrations of ctDNA remains the major challenge for widespread tumor screening and treatment monitoring. Several key technologies have been developed and applied to detect potential micro-metastases and local relapse before clinical and radiological manifestation. This includes several ways of targeted sequencing of known mutations as well as non-targeted, i.e., genome-wide analysis, and fragmentomics[48].
Targeted analysis
Targeted analysis aims to identify tumor-specific mutations or methylations in ctDNA. Techniques include digital PCR (dPCR), BEAMing (beads, emulsions, amplification, magnetics), safe-sequencing system (Safe-SeqS), cancer personalized profiling by deep sequencing (CAPP-Seq), and tagged-amplicon deep sequencing (Tam-Seq). ctDNA can also be analyzed for its relative amount from total cfDNA to serve as a surrogate for tumor burden. This allows risk assessment and staging, as well as early monitoring of therapy response or treatment failure. Tight monitoring can detect mutations as targets or escape mechanisms and supports clinical decision making based on tumor evolution and resistance development. Specific mutations in the EGFR gene in ctDNA allow treatment of NSCLC with a tyrosine kinase inhibitor (TKI)[29,49]. Activating mutations of phosphatidylinositol-3-kinase catalytic subunit alpha (PIK3CA) are used to guide treatment in breast cancer. Resistant tumor cells can escape from therapies by gaining new mutations and clonal selection of the fittest tumor cells. For example, KRAS mutations can develop as a resistance mechanism during EGFR-targeted therapy for patients with CRC[50,51].
DNA methylation is an epigenetic mechanism to regulate gene expression by adding methyl groups to the DNA[52]. In contrast to normal tissue, tumors can show a reversed hyper- and hypomethylation pattern, which can be important for tumor development and progression, as well as for clinical management of patients. Since 2003, typical methylation patterns of tumor DNA have been used to identify ctDNA in blood samples, e.g. MGMT relevant for brain tumors as an actionable target[10,53,54]. More recently, hydroxymethylation profiling with detection of 5-hydroxymethylcytosine (5hmC) as a less known molecular marker of epigenetics was applied in liquid biopsy of lung, pancreatic, and hepatocellular cancer (HCC)[55]. Lung cancer was characterized by a loss of 5hmC, whereas HCC and pancreatic cancer showed disease-specific changes, thus providing information about tumor type and stage.
Non-targeted analysis
Without prior information on specific mutations, non-targeted approaches try to investigate the entire genome, e.g., with whole exome sequencing (WES), whole genome sequencing (WGS), detection of copy number aberrations (CNA), and others[56]. This also allows the detection of subclones evolving under treatment or during natural tumor progression. Unfortunately, low amounts of ctDNA from patients without relapse or metastasis affect sensitivity and utility. Some of these techniques can detect point mutations not previously found in the primary tumor, but with specific value options for treatment and prognosis[57].
Fragmentomics
The size of tumor-derived cfDNA from plasma tends to be shorter than normal background cfDNA[58]. These differences in fragmentation of DNA have been associated with reduced levels of a secreted DNASE1-like nuclease, DNASE1L3, in many tumor types (breast, colorectal, lung, gastric, head and neck non-squamous cell, and liver cancers)[59,60]. Future studies will have to link and translate the biology of circulating DNA and nucleases to include fragment size, end motifs, and jagged (single-stranded) ends into clinical applications in tumor biology[48].
Other types of liquid biopsy: CTCs, EVs, TEPs
Over the last two decades, CTCs entered clinical applications to detect and count them, as well as to determine their genetic profile for prognostics and clinical decision making [Figure 1 and Table 1]. CTCs are usually rare, but they often correlate and contribute to metastatic progression, although only a fraction of CTCs gives rise to metastasis[6]. Despite major advantages of CTCs in diagnostics, prognostics, monitoring, and as guides to therapy choice and delivery schedule, widespread use appears to be still further away. Currently, only a few highly equipped labs supported by major research funds seem to be able to collect and analyze CTCs. A common standard is not established. With CellSearch, the Food and Drug Administration (FDA) and the European Medicines Agency (EMA) approved the first liquid biopsy in 2004[29]. Originally, CellSearch was approved as a diagnostic tool to detect and count CTCs in blood samples to predict outcome (PFS and OS) only in metastatic breast cancer, but it later was expanded to monitor metastatic breast, colorectal, and prostate cancer patients.
miRNA are noncoding, 20-24 nucleotides long RNA molecules derived from just 1% of the whole genome. They are involved in the regulation of stability and translation of mRNA in health and disease. The potential effects of up to 1900 miRNAs are not fully understood. They can be found up- or downregulated in serum, EVs, and CTCs, as well as in urine[61]. Future studies may also include further RNA molecules, such as larger and more stable circular RNA (circRNA). circRNA can serve as an antagonistic sponge for miRNAs and can be involved in gene regulation of tumor cells. However, common standards are needed to be validated, especially since dysregulation of miRNA and circRNA may also be found in inflammatory diseases.
Tumors and normal cells can release small EVs, which contain typical proteins, DNA, and RNA. The intact cell membrane protects the enclosed compounds against degrading enzymes, such as RNAses, from outside the vesicle. Therefore, EVs can be analyzed for the potential markers. Isolation of EVs from blood is the method of choice for most solid tumors. As an exception, CSF to analyze EVs from brain tumors can give better results compared to blood samples for tumors growing close to the ventricles and due to a better signal-to-noise ratio for tumor vs. non-tumor EVs[62].
Blood platelets are derived as anucleated cytoplasmic fragments from megakaryocytes. They can react to activation of membrane receptors and outside-in signaling and have been shown to facilitate metastasis via several mechanisms, including protecting tumor cells within the circulation from immune cells and shear stress, supporting the adhesion to endothelium through adhesion receptors, and releasing of angiogenic and mitogenic growth factors at sites of metastasis. Surprisingly, tumors can alter the RNA profile of platelets, leading to the term tumor educated platelet (TEP)[63]. The mechanism remains to be elucidated. TEPs from lung, brain, and breast cancer patients have been shown to be distinct from those with inflammatory or other diseases[19,64].
CLINICAL STUDIES
Currently, the registry ClinicalTrials.gov from the NIH[65] finds over 900 clinical trials related to the search term “ctDNA” (911 as of 15 March 2022), most of them are ongoing (with 648 “not yet recruiting”, “recruiting”, “enrolling by invitation”, or “active, not recruiting”) and only a minority (95) listed as “completed”, the remaining others being declared as “terminated”, “suspended”, or “unknown status”. Many of these studies are not originally intended to prove the use of ctDNA as a possible measurement of treatment response for new therapy schemes, but they just routinely integrate the ctDNA analysis as an early detection arm for success or failure of the treatment, i.e., as a complement, or when data from tissue biopsy are missing. However, some of the studies can use the potential of early detection of MRD, often months before the clinical manifestation of local relapse or metastasis [Table 3]; this lead time allows an earlier adaptation of the therapy decisions, sometimes with the intent to cure. It indicates the emerging role of ctDNA measurements in clinical settings, which may become the new gold standard, especially when such treatment studies will become new therapy regimes - and then may need or accept ctDNA as necessary control.
Examples of ctDNA studies for screening or monitoring different cancers and stages, as well as treatment response
| Year | Author | Tumor | Method | Findings |
| 2008 | Diehl et al.[15] | CRC | Quantification of ctDNA by patient-specific mutations | Prediction/exclusion of relapse after surgery for at least a year (no long-term follow-up) |
| 2015 | Garcia-Murillas et al.[66] | Early-stage breast cancer | Monitoring of patient-specific mutations by dPCR from cfDNA | Monitor for MRD, prediction of metastatic relapse after initial therapy (pilot study) |
| 2015 | Olsson et al.[67] | Breast cancer | Whole genome sequencing of primary tumor, rearrangements by dPCR in plasma | ctDNA quantity predictive of poor survival |
| 2016 | Donaldson and Park[25,29] | Advanced NSCL | cobas EGFR Mutation Test v2 | First ctDNA-based treatment selection EGFR mutation, exons 18-21 (for Erlotinib) |
| 2017 | Phallen[68] | CRC, breast, lung, ovarian, cancer | TEC-seq of cfDNA | Detection of early stage tumors |
| 2017 | Abbosh et al.[82] | Early-stage NSCLC, TRACERx | ctDNA gene profiling with multiplex-PCR NGS | Identify post-operative relapse |
| 2017 | Ng et al.[70] | CRC | PPS-ctDNA, multiplex-PCR | Monitoring after surgery |
| 2017 | Schøler et al.[71] | CRC | Patient-specific mutations for ctDNA | Postoperative detection of residual disease; prognosis for high risk of relapse; early detection of relapse and response to treatment |
| 2017 | Chaudhuri et al.[72] | NSCLC stage I, II, III | Gene panel with CAPPseq ctDNA | Lead time 5.2 months before clinical recurrence |
| 2018 | Mehra et al.[73] | mCRPC, Prostate cancer | cfDNA concentration | Independent prognostic factor for rPFS and OS in first- and second-line chemotherapy |
| 2018 | Annala et al.[74] | mPRPC | ctDNA | Disruption of TP53, BRCA2 or ATM results in worse outcomes on novel AR targeting |
| 2019 | Coombes et al.[75] | Early-stage breast cancer | Signatera assay, Personalized, 16-plex assays for patients-specific mutations in ctDNA | Lead time 8.9 months (up to 2 years) ahead of clinical relapse) |
| 2019 | Garcia-Murillas et al.[76] | Early-stage breast cancer | Monitoring of patient-specific mutations (mutation tracking) by dPCR from plasma cfDNA | Major “proof of concept” supporting the idea of clinical utility for prediction of relapse to be tested in larger studies |
| 2020 | Moding et al.[77] | NSCLC | ctDNA, CAPP-Seq | Detection of MRD with the prediction of benefit from consolidation immunotherapy |
| 2021 | McDuff et al.[78] | LARC | ctDNA | MRD measured by pre- and postoperative ctDNA predicts outcome for chemoradiation |
| 2021 | Taniguchi et al.[79] | CRC | ctDNA | Testing platform combining a prospective screening registry with two phase 3 interventional studies |
| 2022 | Gale et al.[80] | Early stage NSCLC | Monitoring of patient-specific mutations (48 amplicons) | MRD, identification of patients for further therapy |
| 2022 | Tie et al.[81] | Stage II colon cancer | Tumor-informed personalized sequencing | ctDNA-guided approach to reduce adjuvant chemotherapy without compromising recurrence risk |
Breast cancer
Globally, breast cancer is the most frequent cancer and the leading cause of cancer-related death in women, although most patients can be cured at an early stage. After early surgical removal, additional treatments with chemo-, radio, and/or hormone therapy may further prevent a metastatic relapse. However, overtreatment of cancer-free patients also represents a risk of unnecessary side effects. Therefore, patients should benefit from personalized therapies adjusted for as many additional treatments as required and as few as possible. The challenge to identify MRD with ctDNA analysis from blood samples has been addressed in clinical settings.
In a pilot study in 2015, later complemented with a larger cohort in 2019, Garcia-Murillas and colleagues were able to monitor early breast cancer after initial therapy by detection of ctDNA, which was associated with a high risk of relapse in all breast cancer subtypes[66,75]. Individual somatic mutations were identified by sequencing of DNA from the primary tumors. The same mutations were later tracked by digital PCR (dPCR) in patient’s blood samples. Interestingly, the lead time was 10.7 months (95%CI, 8.1-19.1 months), i.e., long before the clinical manifestation of relapse.
Metastatic breast cancer is only rarely curable at a symptomatic stage. In a retrospective study and long follow-up in 2015, Olsson and colleagues combined whole-genome sequencing of primary tumors with personalized dPCR from plasma ctDNA for quantification of tumor-specific chromosomal rearrangements[67]. With this method, they were able to detect 0.01% of tumor DNA content or one rearranged sequence per 10,000 wild-type sequences. With an average lead time of 11 months (range 0-37 months), they were able to detect metastatic recurrence accurately before its clinical manifestation. In addition, long-term disease-free survivors had undetectable ctDNA. This supported the rationale to evaluate ctDNA in larger studies to monitor early metastasis detection, adjust therapy, and avoid overtreatment.
Mutations in the tumor suppressor protein p53 (TP53) are common among most cancers, although with varying frequencies[82,83]. A high prevalence in a subgroup of breast cancers allowed for monitoring those mutations in ctDNA. Using massively parallel sequencing (MPS), Riva and colleagues detected patient-specific mutations in tumor tissue of non-metastatic triple-negative breast cancer (TNBC)[84]. They tracked these patient-specific TP53 mutations in 10 mL of plasma from different time points: (1) before neoadjuvant chemotherapy (NCT); (2) after one cycle; (3) before surgery; and (4) after surgery. With dPCR of ctDNA, a sensitivity of 75% at baseline was achieved. ctDNA levels correlated not only with tumor burden but also with tumor proliferation rate (tumor grade or mitotic index). During NCT treatment, ctDNA levels dropped in all patients. After surgery, no MRD was detectable. However, a shorter survival was correlated with a slow decrease of ctDNA during NCT[84]. Radovich and colleagues confirmed ctDNA and CTC enumeration above standard clinical parameters as independent factors for the worse outcome, i.e., distant disease-free survival (DDFS), DFS, and OS [Table 4][85].
Examples of clinical trials using ctDNA to validate screening tests or to monitor treatment response
| Year | Study | Tumor | Name | Outcome measures |
| 2014[85] | NCT02101385[86] | TNBC | Randomized controlled trial of genomically directed therapy in patients with TNBC | Comparison of 2-year DFS rate in patients with a genomically directed therapy or standard of care following preoperative chemotherapy |
| 2019 | NCT03934866[87] Observational study (25,000 participants, 55-77 years with smoking history) | Multiple types of cancer (smoking history), lung cancer | SUMMIT - Cancer screening study using GRAIL’s blood test | Validate blood tests for early detection of multiple types of cancers Examine the performance of LDCT screening |
| 2020 | NCT04089631[88] Ongoing interventional clinical trial Phase 3 Drug: Capecitabine | Colon cancer stage II | CIRCULATing tumor DNA-based decision for adjuvant stage II Evaluation (CIRCULATE) | Primary outcome measure: DFS of ctDNApos patients; chemotherapy vs. follow-up Secondary outcome measures: OS in ctDNApos patients with adj. therapy vs. follow-up (5 yrs) DFS in ctDNAneg patients (3 yrs) OS in ctDNAneg patients; Kaplan-Meier (5 yrs) DFS and OS of ctDNApos vs. ctDNAneg (3 and 5 yrs) Site of metastasis (5 yrs); lymph node vs. peritoneal/local recurrence vs. other ctDNApos vs. ctDNAneg |
| 2021 | NCT04931732[89] Observational study | Glioma | circTeloDIAG | Sensitivity and specificity for diagnosis and follow-up |
Colorectal cancer
Colorectal cancer is the second and third most common cancer in women and men, respectively, with almost two million new cases each year[90].
In 2016, Tie and colleagues investigated a prospective cohort of patients after resection of stage II colon cancer[24]. They used massively parallel sequencing to identify patient-specific mutations from primary tumors and designed personalized Safe-SeqS assays for the identified mutations to quantify ctDNA from plasma. The presence of ctDNA in 7.9% of patients not treated with adjuvant chemotherapy was highly associated with recurrence of the disease within the median follow-up of 27 months (79% of patients). In patients who completed chemotherapy, the detection of ctDNA also correlated with a worse outcome. Therefore, ctDNA is useful to identify MRD and the high risk of recurrence in patients with stage II colon carcinoma.
Ng and colleagues developed a patient primary-tumor specific (PPS) assay to detect ctDNA in plasma samples by tracking individual mutations identified from tumor tissue[70]. ctDNA correlated well with clinical treatment outcome: ctDNA was detectable before, but not directly after, surgery. Furthermore, ctDNA was detectable ahead of clinical manifestation of recurrence, indicating its validity to detect metastasis earlier than established methods of imaging and other markers (CEA).
Schøler and colleagues also showed that ctDNA can be useful for monitoring patients with CRC[71]. The detection of ctDNA after surgery reflected MRD and identified patients with a very high risk of relapse. Follow-ups of three years allowed early detection of relapse with a lead time of 9.4 months compared to standard medical imaging, as well as monitoring additional treatment responses. In contrast to CEA levels, preoperative ctDNA detection rates correlated well with the stage of disease.
For example, CIRCULATing tumor DNA-based decision for adjuvant stage II Evaluation (CIRCULATE), as an ongoing multicenter, prospective, randomized, controlled interventional trial (NCT04089631;
Lung cancer
Lung cancer is the leading cause of cancer death (18.4% of total cancer deaths) and, for both sexes combined, the most common cancer (11.6%) with about 2.1 million cases each year[90]. In a seminal paper, Abbosh and colleagues used a phylogenetic approach to track the genetic dynamics of a tumor[69]. They identified predictors of ctDNA in early-stage NSCLC: non-adenocarcinoma histology, necrosis, high proliferative index, and lymphovascular invasion. Chaudhuri and colleagues used a gene panel of 128 genes for CAPP-Seq to predict recurrence with an average lead time of 5.2 months[72]. In 2016, the FDA broadened the approval of the Cobas test (Roche) to be used not only in tissue sections but also as the first liquid biopsy assay for treatment selection to identify specific EGFR mutations in cfDNA of advanced NSCLC patients
The SUMMIT study at the University College London hospitals plans to enroll 25,000 participants 55-77 years of age who have no diagnosis of cancer but a high risk for lung cancer due to a significant smoking history[87]. This study is an ongoing, prospective, observational, cohort study with two major aims: (1) to validate the collaborating company’s (GRAIL, LLC) blood test on cell-free nucleic acids (cfNAs) for early detection of multiple types of cancer; and (2) to deliver low-dose CT (LDCT) screening for lung cancer. Participants will receive at least one low-dose chest CT at baseline. Another possible scan after 12 months will be randomized. The study aims to keep most scans below 1 mSv radiation dose (ultra-low dose) and all scans under 2 mSv. The liquid biopsy will evaluate the test performance, including sensitivity, specificity, and tissue of origin of detected cancers. The performance of the LDCT screening to detect lung cancers will be evaluated in comparison to established measures of risk prediction of lung cancers and other incidental findings. The study started in April 2019 with an estimated primary completion date of August 2023 and an estimated completion date of August 2030[87].
Prostate cancer
Prostate cancer is the second most common non-cutaneous cancer in men worldwide with an estimated 1.3 million new cases each year with more than 350,000 deaths[90]. Prostate cancer contributes to 3.8% of all cancer deaths and is diagnosed in 7.1% of all cancer patients. Mehra and colleagues detected the levels of cfDNA in mCRPC patients in phase 3 clinical studies with first- and second-line chemotherapies[73]. Baseline cfDNA concentration, which is partially derived from the tumor, was an independent prognostic factor in both first- and second-line chemotherapy settings, and it correlated with known prognostic factors, shorter radiological PFS (rPFS), and OS. Higher levels of cfDNA before chemotherapy were associated with more aggressive tumors. A decrease of cfDNA within the first few weeks after initiation of chemotherapy correlated with a benefit for the patient, thus identifying a treatment response. Other studies found that cfDNA can include 15%-20% of ctDNA, depending on tumor stage and tumor burden, but localized prostate cancers typically remain below the threshold for detection of ctDNA, whereas prostate-specific antigen (PSA), a protein marker that is not uniquely an indicator for cancer, is already at a high-risk level[91]. This implies that cfDNA may currently not be feasible to detect and monitor early state or less aggressive tumors. Annala and colleagues[74] investigated cfDNA from mCRPC patients prior to novel AR therapy. Using whole-exome sequencing (WES) with capturing of coding regions of 72 selected genes, they correlated mutations of TP53, BRCA2, or ATM as predictors of worse outcomes on novel AR targeting, thus suggesting liquid biopsy as new guidance in AR-targeted therapy in general practice.
Brain tumors
Cancers of the brain and nervous system represent only 1.6% of all cancers and contribute to 2.5% of all cancer deaths. The critical location within the brain and the lack of major improvements for one of the most devastating cancers, glioblastoma, for over a century represent major challenges. Recently, Eibl and Schneemann published a review on liquid biopsy of primary brain tumors[31]. In contrast to tumors outside of the nervous system, the blood–brain barrier (BBB) may add another challenge for detecting low-level ctDNA in the blood. CSF-although also protected by the BBB-appears to be a much better source due to the normally diminished number of leukocytes as a background source for cfDNA. However, earlier studies were able to confirm specific mutations known from tissue biopsy also in the serum[10,21,92,93], plasma[94-96], or both[21]. In 1991, one of the authors described the very first TP53 mutations in primary medulloblastoma tissue biopsies[82]. This finding supports a model of histologically indistinguishable from primitive neuroectodermal tumors (PNET)[97,98]. Others were unable at that time to detect TP53 mutations in tissue biopsies or in xenografts of human medulloblastoma, except in only one cell line[99]; however, this mutation may have been developed as a selective advantage during cell culture. Similar brain tumor models helped to elucidate and confirm several other oncogenic pathways in human brain tumors[98,100-107]. Meanwhile, TP53 mutations in medulloblastomas are well established and can be used as a prognostic and diagnostic marker: only recently, in 2016[34] and with an update in 2021[108], the World Health Organization (WHO) introduced four new diagnostic groups of this childhood brain tumor based solely on molecular genetic features. The correlation between different biological behavior and personalized risk assessment may allow preventing harmful radiation when not necessary or useful. The first two groups refer to different oncogenic signaling pathways, namely wingless/Integration-1 (WNT)-activated (Group 1) and sonic hedgehog (SHH)-activated (Group 2). WNT is a portmanteau for the Drosophila gene “wingless” (Wg), detected in mutants lacking wings, and the homologous mouse gene, integration 1 (Int-1), which was found earlier to cause tumors by insertional mutagenesis with a retrovirus; SHH refers to the hedgehog gene (hh) found in Drosophila mutants with spikes, reminiscent of a hedgehog (SHH is a vertebrate homolog and named after a character in a video game, Sonic the Hedgehog). WNT-activated medulloblastoma shows the highest five-year survival and a low prevalence of metastatic diseases. SHH-activated medulloblastoma can be further separated into two different subgroups, TP53-mutant or TP53-wildtype. SHH-activated, TP53-mutant occurs primarily in older children and has a very poor prognosis, whereas SHH-activated, TP53-wildtype, which is most common in adolescents and young children, has a good prognosis. The other two groups are non-WNT/non-SHH, Groups 3 and 4, respectively (also known as Groups C and D). Group 3 shows an increased prevalence of metastatic disease with the poorest five-year survival, whereas Group 4 has an increased prevalence of metastatic disease with a moderate five-year survival. TP53 mutations in SHH medulloblastomas are associated with poor survival and treatment failures[34]. Several subgroups have been associated with TP53 and other mutated genes: for WNT-activated, CTNNB1 and APC; for SHH-activated, TP53, PTCH1, SUFU, SMO, MYCN, and GLI2 (methylome); and for non-WNT/non-SHH, MYC, MYCN, PRDM6, and KDM6A (methylome). Since the WHO classification suggests that the diagnosis from molecular profiling of a tissue biopsy is even superior to classical histopathology, at least for brain tumors, it appears reasonable to use ctDNA-based liquid biopsy for monitoring such mutations in brain tumor patients to avoid repeated and risky neurosurgical biopsies[109]. Newer studies successfully used panels of genes. For brain tumors, CSF offers another chance to find ctDNA with a higher sensitivity than plasma or serum[110-114]. ctDNA from CSF even represents the genomic mutations better than plasma; CSF shows an increased sensitivity for putative actionable mutations and CNA (copy number aberrations; EGFR, PTEN, ESR1, IDH1, ERBB2, and FGFR2)[115]. Glioblastoma (glioblastoma multiforme, GBM) represents the most malignant brain tumor. Even after complete surgical removal, the tumor always relapses due to locally infiltrating cells. Chemo- and radiotherapy treatments can help temporarily, but they also trigger the evolution of the tumor to escape all current treatments. Repeated resection and biopsies are generally not indicated. Therefore, a real-time liquid biopsy with ctDNA from CSF offers an alternative at lower risk to monitor the molecular adaptations of the tumor. CSF as a source for ctDNA also provides an additional chance to investigate brain metastases from tumors from outside of the brain better, since they may contribute to the ctDNA in the blood to a lesser extent than metastases from outside of the brain.
ctDNA TESTS
Several ctDNA tests have been developed over the past few years. The spectrum of applications includes early screening, detection of targetable mutations to predict treatment response, MRD, and monitoring after treatment with early detection of resistance [Table 5][132]. After extraction of DNA from plasma, the obtained cfDNA is typically used with qualitative PCR or sequencing methods. Such tests can be highly personalized and target varying numbers of tumor-associated or actionable mutations; they can use a custom-built approach to profiling tumor tissue first for patient- and tumor-specific mutations and later use dPCR techniques to monitor tumor development and treatment response from repeated ctDNA probes.
Examples of ctDNA tests for tumor screening, monitoring, and guiding treatment
| Test | Method | Genes | Cancer type | Comments |
| Bluestar Genomics[116-118] | Hydroxymethylome, NGS, AI | Abnormal genomic/epigenomic signature; 5hmC | Pancreatic cancer (breast and lung cancer) | Screening of patients with new-onset of diabetes, BDD |
| CancerSEEK[26] | ctDNA plus 8 protein markers | ctDNA: 61 Amplicons | Ovary, breast, liver, and others | Sensitivity of about 70% for all 8 cancer types (33%-100% variability) |
| CellMax-LBx[119] | NGS | Mutation profile of 73 genes | Solid tumor | Associated with cancer treatment and tumor response |
| Circulogene[120] | NGS | Approximately 3,000 mutations in > 50 genes | A broad range of tumors | CAP, CLIA |
| Cobas EGFR mutation test v2[29,49] - (Roche) | PCR, actionable EGFR mutations (exons18-21) | cfDNA | Metastatic NSCLC | FDA approved for plasma/liquid biopsy 2016 to identify patients for the first Erlotinib treatment |
| DETECT-A[27] – Thrive (developed into: MCED - Exact Sciences)[121] | Early version of CancerSEEK[26] ctDNA plus protein markers | ctDNA | Lung, ovarian, CRC and others | 1 yr prospective study detecting 26 cancers first in blood |
| FoundationOne Liquid CDx (Roche Foundation Medicine)[122] | NGS | > 300 genes | NSCLC Prostate, ovarian, breast cancer | FDA approved companion diagnostic for treatment |
| Guardant360 CDx[123,124] | NGS | 74 genes | Any advanced solid tumors | FDA approved (for 55 genes) tumor mutation profiling |
| Invitae[125,126] (former ArcherDX) | NGS | ctDNA | Most tumor types | Originally for FFPE, also for ctDNA |
| Oncomine Pan-Cancer Cell-Free Assay[127,128] - ThermoFisher | NGS | cfDNA 52 genes | Various tumors | CE, Europe |
| RaDaR - Inivata[80,129] | NGS | Up to 48 genes | Early stage cancer, NSCL | Predicts early relapse, MRD, BDD |
| Signatera[82,130] - Natera | NGS | ctDNA | A broad range of solid tumors | Custom-built personalized MRD assay; BDD |
| Target Selector - Biocept[131] | NGS | 18 hotspot genes; 10 (breast) and 11 (lung) | Lung and breast cancer | Focus on actionable genes |
| Therascreen PIK3CA (Qiagen)[52] | PCR | 11 activating mutations in exons 7, 9, and 20 of PIK3CA gene | Breast cancer | FDA approved, Identification of patients eligible for PIQRAY (alpelisib) |
Tumor screening
In a large study, the “Detecting cancers Earlier Through Elective mutation-based blood Collection and Testing” (DETECT-A) blood test demonstrated the proof-of-concept to use ctDNA for tumor screening. The DETECT-A test represents an early version of the CancerSEEK test [Table 5] with minor differences, such as using pre-defined thresholds for each DNA and protein biomarker instead of employing artificial intelligence (AI)/machine learning for increasing sensitivity, or for confirming or enhancing specificity[27]. With a cohort of about 10,000 women between 65 and 75 years of age with no history of cancer and one year of follow-up, 26 cancer patients were considered with positive blood testing to be “first detected by blood testing” after confirmation by positron emission tomography-computed tomography (PET-CT): nine lung cancers, six ovarian cancers, and two colorectal cancers - most of them localized or regional. Overall, 14 of the 26 cancers had elevated ctDNA levels, 11 had elevated protein marker levels, and 1 had both elevated ctDNA and protein markers. This test was not designed for regulatory approval, but it addressed fundamental issues of feasibility and safety of multi-cancer blood tests. Indeed, the authors confirmed that a minimally invasive ctDNA-based test was able to safely detect several types of cancer in patients who had no previously known cancer. This allowed early treatment with the intent to cure. The test also produced 101 false positives and 46 false negatives, which still may allow its use as complementary to standard screening methods, although it cannot be used as a stand-alone test. With the use of AI and technology evolution, further improvements appear to be possible.
Tumor monitoring and treatment guidance
Only a few ctDNA tests are FDA approved for guiding treatment choices to identify cancer patients for mutation-specific treatments: Cobas, FoundationOne Liquid CDx, Guardant 360 CDx, and Therascreen PIK3CA [Table 5]. As an example of detecting targetable mutations in cancer patients, the Cobas EGFR mutation test v2 (Roche) aims to detect by PCR from plasma in less than 4 h specific and actionable mutations in the EGFR gene exons 18-21[49]. The FDA approved this test in 2016 for use in liquid biopsy to identify patients for first-line EGFR-targeted therapy (e.g., erlotinib, a tyrosine kinase inhibitor (TKI)) of metastatic NSCLC patients. A similar test was approved earlier for FFPE samples from tumor tissue. In contrast to more recently developing ctDNA tests, the first CTC test approved by the FDA, in 2004, was CellSearch (Veridex). That test uses size, density, electrical properties, and immune surface markers (EpCAM+, Cytokeratins+, and CD45-) for selection and has been cleared for breast, colorectal, and prostate cancer to predict outcome[12].
VISION AND CHALLENGES
As a new milestone in precision medicine, ctDNA-based liquid biopsy has shown its principal utility and safety for an emerging new era of clinical applications. Nevertheless, a few major challenges need to be addressed within the next few years to reach a widespread benefit for cancer patients: the sensitivity of ctDNA diagnostics may increase by technology improvements, thus allowing even earlier detection and, hopefully, easy-to-use multi-cancer screening platforms for at least the most common cancers. A focus will lie on those as well as developing better treatment options including with the intent to cure. Simple tests, such as detecting only actionable mutations, e.g. EGFR exon mutations, may be further developed for not yet used genes, for example, rare and tumor-specific CD44v variant exon combinations[133]. Although many studies describe a significant “lead time” to detect a relapse before any clinical methods or radiologic imaging, one should reflect that any of these clinical methods may also improve over time, e.g., a 3 or 7 Tesla (T) MRI should have a better resolution and detection capacity than a 1.5 T MRI, thus pointing to the uncertainty of comparisons over time. Hundreds of recent and ongoing clinical trials already apply ctDNA methods to monitor new treatments in cancer patients[65,134]. Large multicenter studies will further show how to best standardize and incorporate the use of ctDNA in clinical guidelines. Other arms of liquid biopsy may also be improved in parallel. Living CTCs should be further characterized by not yet applied technologies, such as functional assays with quartz-crystal microbalance (QCM)[135] or atomic force microscopy (AFM), e.g., to check for tumor-specific cell adhesion and pharmacology reactions at the single-molecule level, both applied and pioneered by one of the authors[136-144]. Sharing pharmacological and migratory data on CTCs, and making them findable, accessible, interoperable, and reusable (FAIR), will allow meta-analysis, data integration, and data mining to accelerate such new approaches in liquid biopsy[145]. Some of these experimental technologies may be tested in established models first to allow better reproducibility[146,147]. CellSearch with counting CTCs became clinically validated as the first CTC marker for worse outcomes in early breast cancer[148]. It appears reasonable to combine such CTC tests with ctDNA methods to improve liquid biopsy, perhaps similarly to how CancerSEEK successfully combined ctDNA mutations with protein markers.
CONCLUSION
Depending on tumor type and treatment options, ctDNA has been shown to be a valid and independent biomarker for confirming and serial monitoring of cancer. With its very high specificity, ctDNA can be used to predict the early recurrence of a wide variety of cancers after initial therapy. This enables guidance to earlier and better treatment options, including the intent to cure, but also to prevent patients from unnecessary treatments, when there is no indication for MRD or early detection of resistance development. Numerous larger and ongoing clinical studies with ctDNA as a marker for monitoring MRD or treatment response will increase our knowledge on how to apply the best strategies.
Unfortunately, sensitivity often appears to be too low to use ctDNA for general tumor screenings, and not all patients can be monitored reliably. Further improvement in technology is needed to increase sensitivity and standardization. Although ctDNA appears to be much closer to widespread routine use than the other arms of liquid biopsy (CTCs, EVs, and TEPs), perhaps, a combination of ctDNA data with functional studies on living CTCs can soon close the gap for a faster shift to the new paradigm of liquid biopsy in personalized medicine for the benefit of most cancer patients at any stage of diseases. ctDNA as a valid biomarker is ready to enter clinical routine.
DECLARATIONS
AcknowledgmentsWe thank C. Alix-Panabières for discussion and comments on the manuscript. Since this is such a rapidly emerging field between basic research and clinical applications, we apologize for not being able to include all interesting studies in this review.
Authors’ contributionsMade substantial contributions to conception and design of the study and performed data analysis and interpretation: Eibl RH, Schneemann M
Performed data acquisition, as well as provided administrative, technical, and material support: Eibl, RH, Schneemann M
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2022.
REFERENCES
1. SMART. Servier Med Art. Available from: https://smart.servier.com/ [Last accessed on April 5, 2022].
2. Creative Commons - Attribution 3.0 Unported - CC BY 3.0. Available from: https://creativecommons.org/licenses/by/3.0/ [Last accessed on April 5, 2022].
3. Ashworth A. A case of cancer in which cells similar to those in the tumours were seen in the blood after death. Aust Med J 1869;14:146.
4. Paget S. The distribution of secondary growths in cancer of the breast. The Lancet 1889;133:571-3.
6. Fidler IJ. Biological behavior of malignant melanoma cells correlated to their survival in vivo. Cancer Res 1975;35:218-24.
7. Leon SA, Shapiro B, Sklaroff DM, Yaros MJ. Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res 1977;37:646-50.
8. Lo Y, Wainscoat J, Gillmer M, Patel P, Sampietro M, Fleming K. Prenatal sex determination by DNA amplification from maternal peripheral blood. The Lancet 1989;334:1363-5.
9. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature 2001;414:105-11.
10. Balaña C, Ramirez JL, Taron M, et al. O6-methyl-guanine-DNA methyltransferase methylation in serum and tumor DNA predicts response to 1,3-bis(2-chloroethyl)-1-nitrosourea but not to temozolamide plus cisplatin in glioblastoma multiforme. Clin Cancer Res 2003;9:1461-8.
11. Allard WJ, Matera J, Miller MC, et al. Tumor cells circulate in the peripheral blood of all major carcinomas but not in healthy subjects or patients with nonmalignant diseases. Clin Cancer Res 2004;10:6897-904.
12. Cristofanilli M, Budd GT, Ellis MJ, et al. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N Engl J Med 2004;351:781-91.
13. Diehl F, Li M, Dressman D, et al. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc Natl Acad Sci USA 2005;102:16368-73.
14. Maheswaran S, Sequist LV, Nagrath S, et al. Detection of mutations in EGFR in circulating lung-cancer cells. N Engl J Med 2008;359:366-77.
15. Diehl F, Schmidt K, Choti MA, et al. Circulating mutant DNA to assess tumor dynamics. Nat Med 2008;14:985-90.
16. Cohen SJ, Punt CJ, Iannotti N, et al. Relationship of circulating tumor cells to tumor response, progression-free survival, and overall survival in patients with metastatic colorectal cancer. J Clin Oncol 2008;26:3213-21.
17. de Bono JS, Scher HI, Montgomery RB, et al. Circulating tumor cells predict survival benefit from treatment in metastatic castration-resistant prostate cancer. Clin Cancer Res 2008;14:6302-9.
18. Pantel K, Alix-Panabières C. Circulating tumour cells in cancer patients: challenges and perspectives. Trends Mol Med 2010;16:398-406.
19. Calverley DC, Phang TL, Choudhury QG, et al. Significant downregulation of platelet gene expression in metastatic lung cancer. Clin Transl Sci 2010;3:227-32.
20. Dawson SJ, Tsui DW, Murtaza M, et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med 2013;368:1199-209.
21. Bettegowda C, Sausen M, Leary RJ, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med 2014;6:224ra24.
22. Sullivan JP, Nahed BV, Madden MW, et al. Brain tumor cells in circulation are enriched for mesenchymal gene expression. Cancer Discov 2014;4:1299-309.
23. Mazel M, Jacot W, Pantel K, et al. Frequent expression of PD-L1 on circulating breast cancer cells. Mol Oncol 2015;9:1773-82.
24. Tie J, Wang Y, Tomasetti C, et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci Transl Med 2016;8:346ra92.
25. Donaldson J, Park BH. Circulating tumor DNA: measurement and clinical utility. Annu Rev Med 2018;69:223-34.
26. Cohen JD, Li L, Wang Y, et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 2018;359:926-30.
27. Lennon AM, Buchanan AH, Kinde I, et al. Feasibility of blood testing combined with PET-CT to screen for cancer and guide intervention. Science 2020;369:eabb9601.
29. Research C for DE and. cobas EGFR Mutation Test v2. FDA 2018.
30. Alix-Panabières C, Pantel K. Liquid biopsy: from discovery to clinical application. Cancer Discov 2021;11:858-73.
32. Feng D, Lv J, Li K, et al. CircZNF609 promotes bladder cancer progression and inhibits cisplatin sensitivity via miR-1200/CDC25B pathway. 2022.
34. Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol 2016;131:803-20.
35. Müller NL, Bergin CJ, Miller RR, Ostrow DN. Seeding of malignant cells into the needle track after lung and pleural biopsy. Can Assoc Radiol J 1986;37:192-4.
36. Joosse SA, Beyer B, Gasch C, et al. Tumor-associated release of prostatic cells into the blood after transrectal ultrasound-guided biopsy in patients with histologically confirmed prostate cancer. Clin Chem 2020;66:161-8.
37. Eslami-S Z, Cortés-Hernández LE, Cayrefourcq L, Alix-Panabières C. The different facets of liquid biopsy: a kaleidoscopic view. Cold Spring Harb Perspect Med 2020;10:a037333.
38. Kuske A, Gorges TM, Tennstedt P, et al. Improved detection of circulating tumor cells in non-metastatic high-risk prostate cancer patients. Sci Rep 2016;6:39736.
39. Ramirez JM, Fehm T, Orsini M, et al. Prognostic relevance of viable circulating tumor cells detected by EPISPOT in metastatic breast cancer patients. Clin Chem 2014;60:214-21.
40. Cayrefourcq L, De Roeck A, Garcia C, et al. S100-EPISPOT: a new tool to detect viable circulating melanoma cells. Cells 2019;8:755.
41. Mazard T, Cayrefourcq L, Perriard F, et al. Clinical relevance of viable circulating tumor cells in patients with metastatic colorectal cancer: the COLOSPOT prospective study. Cancers (Basel) 2021;13:2966.
42. Garrel R, Mazel M, Perriard F, et al. Circulating tumor cells as a prognostic factor in recurrent or metastatic head and neck squamous cell carcinoma: the CIRCUTEC prospective study. Clin Chem 2019;65:1267-75.
43. Denève E, Riethdorf S, Ramos J, et al. Capture of viable circulating tumor cells in the liver of colorectal cancer patients. Clin Chem 2013;59:1384-92.
44. Cayrefourcq L, Mazard T, Joosse S, et al. Establishment and characterization of a cell line from human circulating colon cancer cells. Cancer Res 2015;75:892-901.
45. Alix-Panabières C, Pantel K. Circulating tumor cells: liquid biopsy of cancer. Clin Chem 2013;59:110-8.
46. Koch C, Kuske A, Joosse SA, et al. Characterization of circulating breast cancer cells with tumorigenic and metastatic capacity. EMBO Mol Med 2020;12:e11908.
47. Neuberger EWI, Sontag S, Brahmer A, et al. Physical activity specifically evokes release of cell-free DNA from granulocytes thereby affecting liquid biopsy. Clin Epigenetics 2022;14:29.
48. Lo YMD, Han DSC, Jiang P, Chiu RWK. Epigenetics, fragmentomics, and topology of cell-free DNA in liquid biopsies. Science 2021;372:eaaw3616.
49. Kwapisz D. The first liquid biopsy test approved. Is it a new era of mutation testing for non-small cell lung cancer? Ann Transl Med 2017;5:46.
50. Misale S, Yaeger R, Hobor S, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 2012;486:532-6.
51. Diaz LA Jr, Williams RT, Wu J, et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 2012;486:537-40.
52. André F, Ciruelos E, Rubovszky G, et al. SOLAR-1 Study Group. Alpelisib for PIK3CA-mutated, hormone receptor-positive advanced breast cancer. N Engl J Med 2019;380:1929-40.
53. Preuss I, Eberhagen I, Haas S, et al. O6-methylguanine-DNA methyltransferase activity in breast and brain tumors. Int J Cancer 1995;61:321-6.
54. Preuss I, Haas S, Eichhorn U, et al. Activity of the DNA repair protein O6-methylguanine-DNA methyltransferase in human tumor and corresponding normal tissue. Cancer Detect Prev 1996;20:130-6.
55. Song CX, Yin S, Ma L, et al. 5-Hydroxymethylcytosine signatures in cell-free DNA provide information about tumor types and stages. Cell Res 2017;27:1231-42.
56. Heitzer E, Haque IS, Roberts CES, Speicher MR. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat Rev Genet 2019;20:71-88.
57. Ignatiadis M, Sledge GW, Jeffrey SS. Liquid biopsy enters the clinic - implementation issues and future challenges. Nat Rev Clin Oncol 2021;18:297-312.
58. Jiang P, Chan CW, Chan KC, et al. Lengthening and shortening of plasma DNA in hepatocellular carcinoma patients. Proc Natl Acad Sci USA 2015;112:E1317-25.
59. Serpas L, Chan RWY, Jiang P, et al. Dnase1l3 deletion causes aberrations in length and end-motif frequencies in plasma DNA. Proc Natl Acad Sci USA 2019;116:641-9.
60. Jiang P, Sun K, Peng W, et al. Plasma DNA end-motif profiling as a fragmentomic marker in cancer, pregnancy, and transplantation. Cancer Discov 2020;10:664-73.
61. Kitano Y, Aoki K, Ohka F, et al. Urinary microRNA-based diagnostic model for central nervous system tumors using nanowire scaffolds. ACS Appl Mater Interfaces 2021;13:17316-29.
62. Chen WW, Balaj L, Liau LM, et al. BEAMing and droplet digital PCR analysis of mutant IDH1 mRNA in glioma patient serum and cerebrospinal fluid extracellular vesicles. Mol Ther Nucleic Acids 2013;2:e109.
63. Best MG, Sol N, Kooi I, et al. RNA-Seq of tumor-educated platelets enables blood-based pan-cancer, multiclass, and molecular pathway cancer diagnostics. Cancer Cell 2015;28:666-76.
64. Best MG, Wesseling P, Wurdinger T. Tumor-educated platelets as a noninvasive biomarker source for cancer detection and progression monitoring. Cancer Res 2018;78:3407-12.
65. Home - ClinicalTrials. gov. Available from: https://www.clinicaltrials.gov/ [Last accessed on 27 Jul 2022].
66. Garcia-Murillas I, Schiavon G, Weigelt B, et al. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Sci Transl Med 2015;7:302ra133.
67. Olsson E, Winter C, George A, et al. Serial monitoring of circulating tumor DNA in patients with primary breast cancer for detection of occult metastatic disease. EMBO Mol Med 2015;7:1034-47.
68. Phallen J, Sausen M, Adleff V, et al. Direct detection of early-stage cancers using circulating tumor DNA. Sci Transl Med 2017;9:eaan2415.
69. Abbosh C, Birkbak NJ, Wilson GA, et al. The TRACERx consortium; The PEACE consortium. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature 2017;545:446-51.
70. Ng SB, Chua C, Ng M, et al. Individualised multiplexed circulating tumour DNA assays for monitoring of tumour presence in patients after colorectal cancer surgery. Sci Rep 2017;7:40737.
71. Schøler LV, Reinert T, Ørntoft MW, et al. Clinical implications of monitoring circulating tumor DNA in patients with colorectal cancer. Clin Cancer Res 2017;23:5437-45.
72. Chaudhuri AA, Chabon JJ, Lovejoy AF, et al. Early detection of molecular residual disease in localized lung cancer by circulating tumor DNA profiling. Cancer Discov 2017;7:1394-403.
73. Mehra N, Dolling D, Sumanasuriya S, et al. Plasma cell-free DNA concentration and outcomes from taxane therapy in metastatic castration-resistant prostate cancer from two phase III trials (FIRSTANA and PROSELICA). European Urology 2018;74:283-91.
74. Annala M, Vandekerkhove G, Khalaf D, et al. Circulating tumor DNA genomics correlate with resistance to abiraterone and enzalutamide in prostate cancer. Cancer Discov 2018;8:444-57.
75. Coombes RC, Page K, Salari R, et al. Personalized detection of circulating tumor DNA antedates breast cancer metastatic recurrence. Clin Cancer Res 2019;25:4255-63.
76. Garcia-Murillas I, Chopra N, Comino-Méndez I, et al. Assessment of molecular relapse detection in early-stage breast cancer. JAMA Oncol 2019;5:1473-8.
77. Moding EJ, Liu Y, Nabet BY, et al. Circulating tumor DNA dynamics predict benefit from consolidation immunotherapy in locally advanced non-small cell lung cancer. Nat Cancer 2020;1:176-83.
78. McDuff SGR, Hardiman KM, Ulintz PJ, et al. Circulating tumor DNA predicts pathologic and clinical outcomes following neoadjuvant chemoradiation and surgery for patients with locally advanced rectal cancer. JCO Precis Oncol 2021;5:PO.
79. Taniguchi H, Nakamura Y, Kotani D, et al. CIRCULATE-Japan: circulating tumor DNA-guided adaptive platform trials to refine adjuvant therapy for colorectal cancer. Cancer Sci 2021;112:2915-20.
80. Gale D, Heider K, Ruiz-Valdepenas A, et al. Residual ctDNA after treatment predicts early relapse in patients with early-stage non-small cell lung cancer. Ann Oncol 2022;33:500-10.
81. Tie J, Cohen JD, Lahouel K, et al. DYNAMIC Investigators. Circulating tumor DNA analysis guiding adjuvant therapy in stage II colon cancer. N Engl J Med 2022;386:2261-72.
82. Ohgaki H, Eibl RH, Wiestler OD, Yasargil MG, Newcomb EW, Kleihues P. p53 mutations in nonastrocytic human brain tumors. Cancer Res 1991;51:6202-5.
83. Zhukova N, Ramaswamy V, Remke M, et al. Subgroup-specific prognostic implications of TP53 mutation in medulloblastoma. J Clin Oncol 2013;31:2927-35.
84. Riva F, Bidard FC, Houy A, et al. Patient-specific circulating tumor DNA detection during neoadjuvant chemotherapy in triple-negative breast cancer. Clin Chem 2017;63:691-9.
85. Radovich M, Jiang G, Hancock BA, et al. Association of circulating tumor DNA and circulating tumor cells after neoadjuvant chemotherapy with disease recurrence in patients with triple-negative breast cancer: preplanned secondary analysis of the BRE12-158 randomized clinical trial. JAMA Oncol 2020;6:1410-5.
86. MD BS. A Phase II Randomized controlled trial of genomically directed therapy after preoperative chemotherapy in patients with triple negative breast cancer: hoosier oncology group BRE12-158. clinicaltrials.gov 2020. Available from: https://clinicaltrials.gov/ct2/show/NCT02101385 [Last accessed on 2 Aug 2022]
87. University College, London. The SUMMIT study: cancer screening study with or without low dose lung CT to validate a multi-cancer early detection test. clinicaltrials.gov; 2021. Available from: https://www.clinicaltrials.gov/ct2/show/NCT03934866 [Last accessed on 28 Jul 2022].
88. Technische Universität Dresden. Circulating Tumour DNA Based Decision for Adjuvant Treatment in Colon Cancer Stage II Evaluation (CIRCULATE) AIO-KRK-0217. clinicaltrials.gov; 2020. Available from: https://clinicaltrials.gov/ct2/show/NCT04089631 [Last accessed on 2 Aug 2022].
89. Hospices Civils de Lyon. The circTeloDIAG: a new approach of liquid biopsy for the diagnosis and follow-up of patients with glioma tumor. clinicaltrials.gov; 2021. Available from: https://clinicaltrials.gov/ct2/show/NCT04931732 [Last accessed on 2 Aug 2022]
90. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018;68:394-424.
91. Hennigan ST, Trostel SY, Terrigino NT, et al. Low abundance of circulating tumor DNA in localized prostate cancer. JCO Precis Oncol 2019;3:PO.19.00176.
92. Lavon I, Refael M, Zelikovitch B, Shalom E, Siegal T. Serum DNA can define tumor-specific genetic and epigenetic markers in gliomas of various grades. Neuro Oncol 2010;12:173-80.
93. Majchrzak-Celińska A, Paluszczak J, Kleszcz R, et al. Detection of MGMT, RASSF1A, p15INK4B, and p14ARF promoter methylation in circulating tumor-derived DNA of central nervous system cancer patients. J Appl Genet 2013;54:335-44.
94. Boisselier B, Gállego Pérez-Larraya J, Rossetto M, et al. Detection of IDH1 mutation in the plasma of patients with glioma. Neurology 2012;79:1693-8.
95. Schwaederle M, Chattopadhyay R, Kato S, et al. Genomic alterations in circulating tumor DNA from diverse cancer patients identified by next-generation sequencing. Cancer Res 2017;77:5419-27.
96. Weaver KD, Grossman SA, Herman JG. Methylated tumor-specific DNA as a plasma biomarker in patients with glioma. Cancer Invest 2006;24:35-40.
97. Eibl RH, Wiestler OD. Induction of primitive neuroectodermal tumors following retrovirus-mediated transfer of SV40 large T antigen into neural transplants. Zülch symposium on growth control and neoplastic transformation in the brain. Goslar, Germany. Rockledge, U.S.A.: Dustri; 1991. pp. 248–9.
98. Eibl RH, Kleihues P, Jat PS, Wiestler OD. A model for primitive neuroectodermal tumors in transgenic neural transplants harboring the SV40 large T antigen. Am J Pathol 1994;144:556-64.
99. Saylors RL, Sidransky D, Friedman HS, et al. Infrequent p53 gene mutations in medulloblastomas. Cancer Res 1991;51:4721-3.
100. Kleihues P, Ohgaki H, Eibl RH, et al. Type and frequency of p53 mutations in tumors of the nervous system and its coverings. Recent Results Cancer Res 1994;135:25-31.
101. Louis DN, von Deimling A, Chung RY, et al. Comparative study of p53 gene and protein alterations in human astrocytic tumors. J Neuropathol Exp Neurol 1993;52:31-8.
102. Ohgaki H, Eibl RH, Schwab M, et al. Mutations of the p53 tumor suppressor gene in neoplasms of the human nervous system. Mol Carcinog 1993;8:74-80.
103. von Deimling A, Eibl RH, Ohgaki H, et al. p53 mutations are associated with 17p allelic loss in grade II and grade III astrocytoma. Cancer Res 1992;52:2987-90.
104. Wiestler OD, Brüstle O, Eibl RH, Radner H, Aguzzi A, Kleihues P. Retrovirus-mediated oncogene transfer into neural transplants. Brain Pathol 1992;2:47-59.
105. Wiestler OD, Brüstle O, Eibl RH, et al. A new approach to the molecular basis of neoplastic transformation in the brain. Neuropathol Appl Neurobiol 1992;18:443-53.
106. Wiestler OD, Brüstle O, Eibl RH, Radner H, Aguzzi A, Kleihues P. Oncogene transfer into the brain. Molecular Neuro-oncology and Its Impact on the Clinical Management of Brain Tumors 1994:5-66.
107. Wiestler OD, Aguzzi A, Brüstle O, Eibl R, Radner H, Kleihues P. Oncogene complementation in transgenic neural transplants. Neuropathology 1990;4:304-9.
108. Louis DN, Perry A, Wesseling P, et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol 2021;23:1231-51.
109. Eibl RH, Schneemann M. Liquid biopsy for monitoring medulloblastoma. Extracell Vesicles Circ Nucleic Acids 2022;3:263-74.
110. Martínez-Ricarte F, Mayor R, Martínez-Sáez E, et al. Molecular diagnosis of diffuse gliomas through sequencing of cell-free circulating tumor DNA from cerebrospinal fluid. Clin Cancer Res 2018;24:2812-9.
111. Miller AM, Shah RH, Pentsova EI, et al. Tracking tumour evolution in glioma through liquid biopsies of cerebrospinal fluid. Nature 2019;565:654-8.
112. Mouliere F, Mair R, Chandrananda D, et al. Detection of cell-free DNA fragmentation and copy number alterations in cerebrospinal fluid from glioma patients. EMBO Mol Med 2018;10:e9323.
113. Pan Y, Long W, Liu Q. Current advances and future perspectives of cerebrospinal fluid biopsy in midline brain malignancies. Curr Treat Options Oncol 2019;20:88.
114. Wang Y, Springer S, Zhang M, et al. Detection of tumor-derived DNA in cerebrospinal fluid of patients with primary tumors of the brain and spinal cord. Proc Natl Acad Sci USA 2015;112:9704-9.
115. De Mattos-Arruda L, Mayor R, Ng CKY, et al. Cerebrospinal fluid-derived circulating tumour DNA better represents the genomic alterations of brain tumours than plasma. Nat Commun 2015;6:8839.
116. Guler GD, Ning Y, Ku CJ, et al. Detection of early stage pancreatic cancer using 5-hydroxymethylcytosine signatures in circulating cell free DNA. Nat Commun 2020;11:5270.
117. Haan D, Bergamaschi A, Guler GD, et al. Validation of a pancreatic cancer detection test in new-onset diabetes using cell-free DNA 5-Hydroxymethylation signatures, 2021. Available from: https://www.medrxiv.org/content/10.1101/2021.12.27.21268450v1 [Last accessed on 27 Jul 2022].
118. Bluestar Genomics. Liquid biopsy cancer detection. BlueStar Genomics. Available from: https://www.bluestargenomics.com/ [Last accessed on 27 Jul 2022].
119. The first blood test for colon cancer screening and prevention. Available from: https://cellmaxlife.in/ [Last accessed on 29 Jul 2022].
120. Home. Circulogene. Available from: https://circulogene.com/ [Last accessed on 27 Jul 2022].
121. Home. Exact Sciences. Available from: https://www.exactsciences.com/ [Last accessed on 27 Jul 2022].
122. Foundation Medicine. A World-leading Molecular Insights Company. Available from: https://www.foundationmedicine.com/ [Last accessed on 27 Jul 2022].
123. Kim ST, Banks KC, Lee SH, et al. Prospective feasibility study for using cell-free circulating tumor DNA-guided therapy in refractory metastatic solid cancers: an interim analysis. JCO Precis Oncol 2017;1:PO.16.00059.
124. Liquid Biopsy For All Advanced Solid Tumors. Guardant360® CDx. Available from: https://guardant360cdx.com/ [Last accessed on 27 Jul 2022].
125. Zheng Z, Liebers M, Zhelyazkova B, et al. Anchored multiplex PCR for targeted next-generation sequencing. Nat Med 2014;20:1479-84.
126. Democratizing precision oncology. ArcherDX. Available from: https://archerdx.com/ [Last accessed on 27 Jul 2022].
127. So MK, Park JH, Kim JW, Jang JH. Analytical validation of a pan-cancer panel for cell-free assay for the detection of. EGFR 2021;11:1022.
128. Scientific TF. Oncomine next-generation sequencing solutions for precision oncology research. Available from: https://www.oncomine.com [Last accessed on 27 Jul 2022].
129. Welcome to Inivata. A Leader in Liquid Biopsy. Cancer Care Treatment. Available from: https://www.inivata.com/ [Last accessed on 27 Jul 2022].
130. Natera: A global leader in cfDNA testing. Natera. Available from: https://www.natera.com/ [Last accessed on 27 Jul 2022].
131. Biocept – Advancing Diagnostics to Improve Cancer Treatments. Available from: https://biocept.com/ [Last accessed on 27 Jul 2022].
132. van der Leest P, Ketelaar EM, van Noesel CJM, et al. Dutch national round robin trial on plasma-derived circulating cell-free DNA extraction methods routinely used in clinical pathology for molecular tumor profiling. Clin Chem 2022;68:963-72.
133. Eibl RH, Pietsch T, Moll J, et al. Expression of variant CD44 epitopes in human astrocytic brain tumors. J Neurooncol 1995;26:165-70.
134. Home. Australian Clinical Trials. Available from: https://www.australianclinicaltrials.gov.au/ [Last accessed on 27 Jul 2022].
135. Pax M, Rieger J, Eibl RH, Thielemann C, Johannsmann D. Measurements of fast fluctuations of viscoelastic properties with the quartz crystal microbalance. Analyst 2005;130:1474-7.
136. Eibl RH, Benoit M. Molecular resolution of cell adhesion forces. IEE Proc Nanobiotechnol 2004;151:128-32.
137. Eibl RH, Moy VT. AFM-based adhesion measurements of single receptor-ligand bonds on living cells. In: Pandalai SG, editor. Recent research developments in biophysics. Trivandrum: Transworld Research Network; 2004:235–46. Available from: https://pascal-francis.inist.fr/vibad/index.php?action=getRecordDetail&idt=17751719 [Last accessed on 27 Jul 2022].
138. Eibl RH, Moy VT. Atomic force microscopy measurements of protein–ligand interactions on living cells. Methods Mol Biol 2005;305:439-50.
139. Eibl RH. Direct force measurements of receptor–ligand interactions on living cells. In: Bhushan B, Fuchs H, editors. Applied Scanning Probe Methods XII: Characterization. Berlin, Heidelberg: Springer; 2009. pp. 1-31.
140. Eibl RH. Cell adhesion receptors studied by AFM-Based single-molecule force spectroscopy. In: Bhushan B, editor. Scanning Probe Microscopy in Nanoscience and Nanotechnology 2. Berlin, Heidelberg: Springer; 2011. pp. 197-215.
141. Eibl RH. Single-molecule studies of integrins by AFM-based force spectroscopy on living cells. In: Bhushan B, editor. Scanning Probe Microscopy in Nanoscience and Nanotechnology 3. Berlin, Heidelberg: Springer; 2013. pp. 137–69.
142. Eibl RH. Comment on "A method to measure cellular adhesion utilizing a polymer micro-cantilever". Appl Phys Lett 2014;104:236103.
143. Eibl RH, Moy VT. Atomic force microscopy measurements of protein-ligand interactions on living cells. In: Ulrich Nienhaus G, editor. Protein-Ligand Interactions: Methods and Applications. Totowa, NJ: Humana Press; 2005. pp. 439-49.
144. Eibl RH. First measurement of physiologic VLA-4 activation by SDF-1 at the single-molecule level on a living cell. In: Hinterdorfer P, Schütz G, Pohl P, editors. Proceedings of the VIII. Linz Winter Workshop 2006: Advances in Single Molecule Research for Biology and Nanoscience. Linz: Trauner; 2006. pp. 40-3.
145. Gonzalez-Beltran AN, Masuzzo P, Ampe C, et al. Community standards for open cell migration data. Gigascience 2020;9:giaa041.
146. Wiestler OD, Aguzzi A, Schneemann M, Eibl R, von Deimling A, Kleihues P. Oncogene complementation in fetal brain transplants. Cancer Res 1992;52:3760-7.
147. Radner H, el-Shabrawi Y, Eibl RH, et al. Tumor induction by ras and myc oncogenes in fetal and neonatal brain: modulating effects of developmental stage and retroviral dose. Acta Neuropathol 1993;86:456-65.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Eibl RH, Schneemann M. Cell-free DNA as a biomarker in cancer. Extracell Vesicles Circ Nucleic Acids 2022;3:195-215. http://dx.doi.org/10.20517/evcna.2022.20
AMA Style
Eibl RH, Schneemann M. Cell-free DNA as a biomarker in cancer. Extracellular Vesicles and Circulating Nucleic Acids. 2022; 3(3): 195-215. http://dx.doi.org/10.20517/evcna.2022.20
Chicago/Turabian Style
Eibl, Robert H., Markus Schneemann. 2022. "Cell-free DNA as a biomarker in cancer" Extracellular Vesicles and Circulating Nucleic Acids. 3, no.3: 195-215. http://dx.doi.org/10.20517/evcna.2022.20
ACS Style
Eibl, RH.; Schneemann M. Cell-free DNA as a biomarker in cancer. Extracell. Vesicles. Circ. Nucleic. Acids. 2022, 3, 195-215. http://dx.doi.org/10.20517/evcna.2022.20
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 27 clicks
Cite This Article 27 clicks

 Like This Article 11
likes
Like This Article 11
likes







Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.