
Expanded knowledge of cell-free DNA biology: potential to broaden the clinical utility

 , ...
, ... Abstract
Noninvasive sampling of an individual’s body fluids is an easy means to capture circulating cell-free DNA (cfDNA). These small fragments of DNA carry information on the contributing cell’s genome, epigenome, and nuclease content. Analysis of cfDNA for the assessment of genetic risk has already revolutionized clinical practice, and a compendium of increasingly higher-resolution approaches based on epigenetic and fragmentomic cfDNA signatures continues to expand. Profiling cfDNA has unlocked a wealth of molecular information that can be translated to the clinic. This review covers the biological characteristics of cfDNA, recent advances in liquid biopsy and the clinical utility of cfDNA.
Keywords
INTRODUCTION
Invasive diagnostic tests, such as tissue biopsies, are limited by procedure-associated risks and sometimes sampling difficulties. There is a strong need for noninvasive or minimally invasive biomarkers that do not rely on targeted sampling and expedite the often lengthy process of identifying disease. Cell-free DNA (cfDNA) has emerged as a vital biomarker for detecting and monitoring disease. Assessment of cfDNA has provided new opportunities to noninvasively obtain information from source tissues of interest. The use of cfDNA has been explored in different fields and implemented to varying extents. In obstetrics, for example, multiple large-scale clinical studies have successfully used cfDNA for prenatal screening of certain genetic conditions, underscoring the clinical value of cfDNA analysis. Specifically, common fetal aneuploidy screening is of high clinical relevance for pregnancy management. In oncology, the use of cfDNA for early cancer detection, prognosis, and treatment is under particularly extensive investigation. Moreover, there is a rising interest in broadening the diagnostic scope of cfDNA both in terms of disease and methodology. In this review, we have summarized current understandings of cfDNA and the clinical utility of cfDNA in different medical fields.
Cell-free DNA
Cell-free DNA (cfDNA) is ubiquitous in human body fluids, including blood, urine, cerebrospinal fluid, sputum, ascites and pleural effusion[1,2]. Serum and plasma cfDNA have been extensively studied. It is generally accepted that cfDNA in the blood of healthy individuals is derived primarily from apoptotic hematopoietic cells[3,4]. Hematopoietic cell lineages have a fast turnover rate and short half-life[5]. In line with this, studies have shown that the main cellular origin of cfDNA is from hematopoietic cells[3,6]. The fragment size distribution of cfDNA is referred to as the apoptotic ladder because it matches the progression of nucleosome units with successive peaks at ~167 bp corresponding to the length of DNA wrapped around a mononucleosome (147 bp) plus the linker regions (20 bp)[7,8], at ~330 bp for the di-nucleosome and at ~500 bp for the tri-nucleosome[9-11]. DNA fragments below 167 bp exhibit a series of smaller peaks at a periodicity of ~10 bp, likely reflecting the DNA helical repeat and cleavage at groove regions where DNA bends sharply around the nucleosome[12,13] [Figure 1]. Other types of cfDNA release mechanisms have also been suggested under certain pathological conditions[14]. For instance, the presence of DNA fragments over 10 kilobases in cancer patients may be indicative of necrosis[10]. NETosis, pyroptosis and active secretion have also been proposed as putative sources of cfDNA[15,16]. In addition to cfDNA release, studies have revealed impaired cfDNA clearance in a number of (patho)physiological states[17-19]. Extracellular nucleases from the deoxyribonuclease (DNase) family are capable of digesting internucleosomal linker regions and enzymatically clearing free and protein-bound DNA[20,21]. CfDNA is then eliminated from circulation through organs, such as the liver, spleen, and kidney[22]. Abnormal DNase activity and insufficient clearance of cellular debris have been associated with elevated cfDNA concentration in patients with autoimmune diseases[23,24] and cancer[25]. Altogether, cfDNA reflects a heterogeneous, complex and dynamic landscape of dying cells in an individual and holds great promise for noninvasive molecular testing.
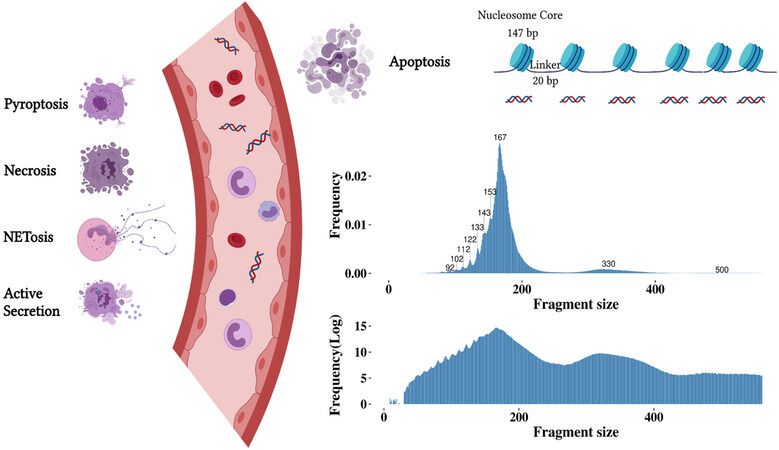
Figure 1. Illustration of cfDNA release mechanisms and the resulting fragment size (in bp) from apoptosis. Different forms of cfDNA release have been suggested, these primarily include apoptosis, pyroptosis, necrosis, the release of neutrophil extracellular traps (NETosis) and active secretion. Apoptosis is considered the primary source of cfDNA, resulting in non-random fragmentation. Other mechanisms of cell death, such as, pyroptosis that is inflammatory-regulated, necrosis that occurs due to accidental cell death, and NETosis that is neutrophil-specific, can also contribute to the release of cfDNA. In addition to cell death, cfDNA might be derived from active cellular secretions. The fragment size distribution of cfDNA shows peaks in sizes below 167 bp and peaks corresponding to nucleosome units. The log-transformed fragment size distribution demonstrates the di- and tri-nucleosome peak.
Cell-free fetal DNA
During pregnancy, fetal DNA fragments are detected in the maternal plasma DNA population. Y chromosomal DNA fragments from a male fetus were observed in the cfDNA of a pregnant individual as early as 1997[26]. It is well-recognized that cell-free fetal DNA (cffDNA) mainly originates from placental trophoblast cells. This conclusion has been drawn from the observation that cffDNA was detectable in anembryonic pregnancies and at early gestational weeks before fetal organ development[27,28]. Moreover, cffDNA shares methylation signatures common to trophoblast cells[29-31] and, in the event of confined placental mosaicism, cffDNA reflects the genotype of the placenta rather than the fetus proper[32,33]. Though the cffDNA concentration increases as gestation advances, the placental contribution to the pool of cfDNA in maternal plasma, often described as the fetal fraction (FF), remains a minor fraction, usually 10%-20% throughout the first and second trimester[34]. The release of cffDNA is tightly linked to placental morphogenesis. Consequently, placental dysfunction can directly affect circulating cffDNA levels. For example, in pregnancies with preeclampsia or at risk of developing preeclampsia, elevated cffDNA levels have been reported[35-37]. Maternal conditions, such as obesity, can lead to lower FF due to higher maternal contributions[38,39]. CffDNA is rapidly removed from maternal circulation after delivery and the estimated half-life of cffDNA clearance is about 1 h[40]. Fragment size of cffDNA is generally shorter than the maternal cfDNA and peaks at around 143 bp[9] [Figure 2]. The shorter size of cffDNA may be related to nucleosome organization and chromatin accessibility in placental tissue[41]. Assay for Transposase-Accessible Chromatin (ATAC) sequencing has revealed that chromatin is more accessible in placental tissue and may indicate that DNA is more likely to be cut close to nucleosome cores resulting in shorter fragments[41]. The basis for investigating the fetal genotype noninvasively is that the entire placental genome is present in the maternal plasma. Lo et al. showed that the proportion of cffDNA in maternal plasma is constant across the genome[9]. The coverage of cffDNA fragments across the genome can, however, fluctuate from region to region, potentially indicative of nucleosomal degradation patterns in the tissue of origin[42,43].
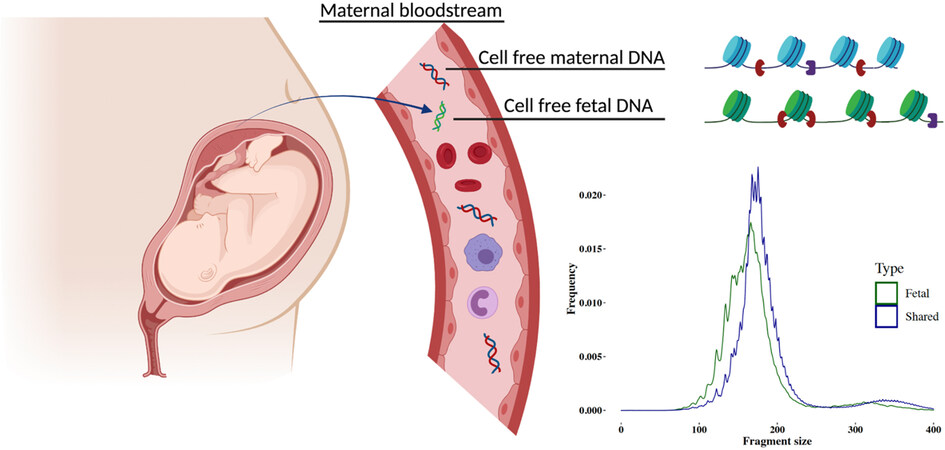
Figure 2. Illustration of maternal plasma DNA and an example of the fragment size (in bp) distribution of fetal-specific DNA (green) and shared (maternal and fetal; blue) DNA. cffDNA has shifted size distribution, being overall shorter than the maternal counterparts.
Circulating tumor DNA
The surge of interest in cfDNA was not only sparked by the discovery of cffDNA, but also by the discovery of circulating tumor DNA (ctDNA) in cancer patients decades ago[44-46]. Studies have shown that genetic alterations observed in ctDNA reflect copy number aberrations and somatic mutations in the primary tumor or multiregional tumor biopsies[47-49]. Analysis of nucleosome maps of plasma cfDNA[50] and methylation signatures[4] in cancer patients have provided further evidence that ctDNA is derived from the malignant cells. It has also been postulated that non-malignant cells that are part of the tumor microenvironment, such as stromal, endothelial, lymphocytes and other immune cells, may contribute to plasma DNA of cancer patients[1,51]. The amount of ctDNA in the circulation, also termed tumor fraction, is likely to be associated with tumor mass/burden, cell turnover and disease stage. Increased amounts of ctDNA were found in cancer patients with advanced stage compared to early stage of diseases[52]. Changes in tumor fractions also reflect treatment responses over the course of therapy[53,54]. Nevertheless, tumor fraction in plasma DNA can remain very low even in metastatic diseases, with ctDNA levels varying across different tumor types and even subtypes[52,55,56]. The anatomical location of the tumor can also influence the amount of ctDNA that is shed into body fluid [Figure 3]. As an example, in patients with bladder cancer, ctDNA is more easily detected in urine compared to in blood[57,58]. Lastly, recent works using paired-end sequencing suggest that malignant cell-derived DNA fragments are in general shorter than those of non-malignant DNA[50,59].
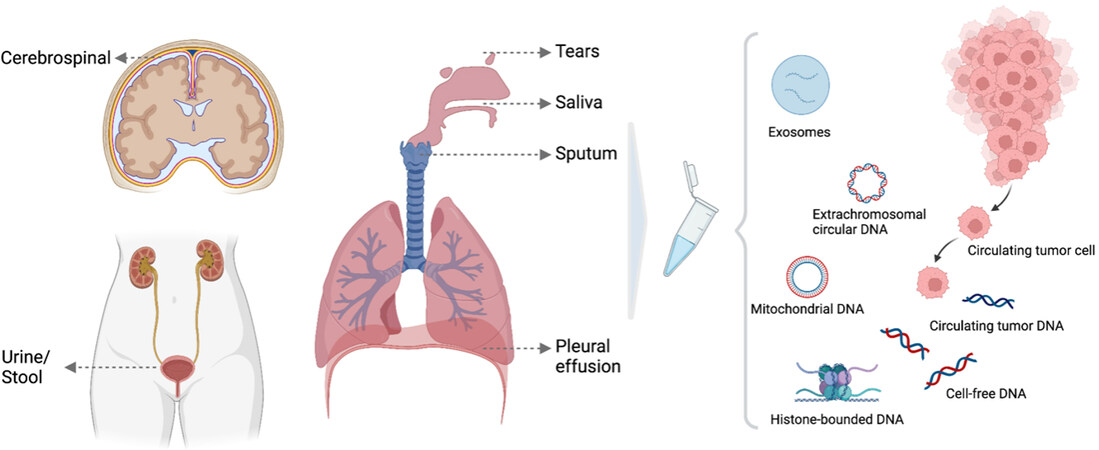
Figure 3. Illustration of ctDNA derived from other body fluids other than blood. Different forms of markers present in biological fluids, including circulating tumor cells, circulating cell-free DNA and circulating tumor DNA, histone-bounded DNA, exosomes, extrachromosomal circular DNA and mitochondrial DNA.
Strategies to analyze plasma cfDNA
Quantitative and qualitative analyses have been developed to interrogate cfDNA. Pre-analytical factors can affect properties of cfDNA. Studies have evaluated the impact of variables, including blood collection, processing, plasma isolation and storage. Recommendations and development of standardized protocols to enhance test performance are advised[60-62]. Quantitative assessment usually involves the extraction of DNA from plasma or other fluids for concentration quantification. CfDNA concentration as a biomarker has been investigated widely in prenatal, cancer and autoimmune disease applications[63-65], as levels of total cfDNA are known to increase under certain conditions and fluctuate in accordance with disease state. In addition to levels of total cfDNA, levels of cffDNA and ctDNA in the plasma of pregnant women and cancer patients, respectively, may also differ from those of controls. By making use of Y chromosome- or placental-specific markers, studies have shown that cffDNA levels in complicated pregnancies appear to be higher[36,66]. Likewise, ctDNA level, measured by a mutation template, has been explored as a biomarker for cancer detection and treatment response[52,67].
While the abundance of total cfDNA and its sub fractions can indicate the presence of (patho)physiological states, a more comprehensive view is obtained by qualitative analyses. Those include the detection of genetic variations, methylation signatures and fragmentation patterns. Next generation sequencing (NGS) technologies have enabled analysis at unprecedented throughput and resolution. The presence of placenta- or tumor-derived DNA in cfDNA pools can be determined through the detection of genetic and epigenetic variations [Figure 4]. The limited absolute number of DNA molecules and low cffDNA or ctDNA concentration against high maternal or non-malignant backgrounds in plasma samples directly affects the signal identification. Though the biological limitations pose obstacles to robust detection of variations, approaches have been developed to address these challenges. One of the first methods that demonstrated sensitive detection of fetal aneuploidies relied on massive parallel sequencing to profile genomic representations of cfDNA[68,69]. Quantifying the statistical deviations of a profile from an external reference profile comprised of normal controls or the deviation of one region from other regions within the same sample allows robust detection of common fetal aneuploidies - an approach that has been validated by multiple large-scale clinical studies[70-72]. The strategy has been shown to capture copy number aberrations within ctDNA as well[73,74]. Deep sequencing of single-nucleotide polymorphisms and, in the case of cancer, somatic point mutations, can also be performed for the quantification of cff- and ct-DNA, respectively. In cancer studies, to compensate for the low number of ctDNA source molecules, larger numbers of mutations are tested to improve sensitivity[75]. Using unique molecular identifiers and analytical error corrections can further facilitate variant or mutation detection[76-78]. A recently developed approach that leverages two or more mutations occurring on the same strand of DNA has further pushed the detection limit. Using a personalized panel, the approach allowed identification of variants with a tumor fraction as low as 1/1,000,000[79]. Another type of genetic alteration - rearrangements - have been tested as part of NGS solutions specific for ctDNA detection, and a high concordance between cfDNA and tumor tissue was obtained for selected gene fusion analyses[80,81].
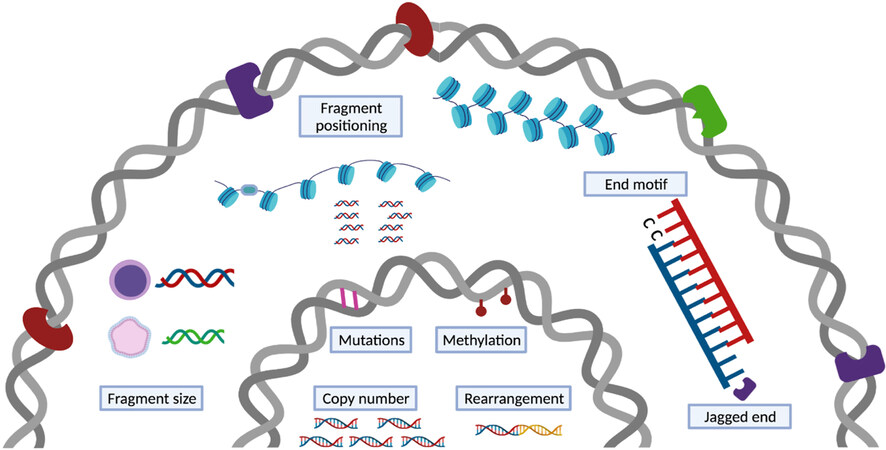
Figure 4. cfDNA carries (epi)genetic and fragmentation signatures. A catalog of genetic and non-genetic signatures that might be used for disease detection. The genetic signatures, including copy number changes, single nucleotide variations, and rearrangements, and epigenetic signatures, mainly methylation, are vastly investigated and used in clinical applications. The non-genetic changes that encompass several dimensions of characteristics resulted from cfDNA release mechanisms are under exploration.
Epigenetic approaches have been exploited extensively in recent years, among which, DNA methylation has been widely adopted for cfDNA analysis in cancer. Shortly after the discovery of tumor- and fetal-derived DNA in plasma samples, tumor and fetal specific methylation patterns were also observed in cfDNA[82,83]. Early in disease development, DNA methylation can exhibit divergent patterns across different tissues/cells[4,6]. Methylation changes thus offer the potential to predict and monitor disease states. Early investigative approaches were limited to DNA methylation marks on individual genes[29,82,84], but have expanded to genome-wide assays[85,86] aimed at improving sensitivity and specificity. For example, approaches that leverage multiple informative CpG markers[87] or methylation haplotype blocks that exhibit highly coordinated methylation across consecutive CpG sites[88] have been developed. The main method to analyze cytosine methylation at single nucleotide resolution is bisulfite conversion. The method makes use of sodium bisulfite treatment, which converts unmethylated cytosine residues to uracil and leaves 5-methylcytosine (5mC) unaffected. This approach has a major drawback in that it causes degradation of the DNA. To overcome this limitation, new methods for methylation analysis are being developed. Such methods include cell-free methylated DNA immunoprecipitation and high-throughput sequencing (cfMeDIP-seq)[89], enzymatic methyl-seq (EM-Seq)[90,91], and ten-eleven translocation (TET)-assisted pyridine borane sequencing (TAPS)[92], which have all been successfully applied for methylation analysis in cfDNA. Furthermore, third-generation sequencing, including single-molecule real-time sequencing by Pacific Biosciences and nanopore sequencing by Oxford Nanopore Technologies, has opened new avenues for cfDNA analysis, enabling simultaneous real-time analysis of DNA sequence as well as nucleotide modifications. Though sequencing fragmented cfDNA is still challenging, proof-of-concept studies have demonstrated the potential of using these new technologies for the development of noninvasive disease management tools[93]. Beyond methylation patterns, chromatin immunoprecipitation and sequencing (ChIP-seq) has been developed to investigate histone modifications, allowing cell-of-origin determination and disease-specific transcriptional programs to be identified[94].
In addition to genetic and epigenetic changes, various analyses that rely on cfDNA fragmentation patterns have come into view. Fragmentation of cfDNA is non-random. It is mechanically related to modes of cell death and nuclease cleavage, and cellularly associated with the nucleosome organization, chromatin structure and gene expression of the tissue of origin. Fragmentation-based signatures or fragmentomics therefore reflect multiple processes that may provide additional markers for clinical use. CfDNA fragment size is one the most thoroughly assessed fragmentation signatures. It has long been recognized from the fragments’ ladder-like patterns that cfDNA bears a signature of caspase-dependent apoptosis[7,10]. More recent data has shown size differences between contributing tissues. Even though the exact cause of size differences between fragments from different tissues remains to be clarified, fragment size patterns have been leveraged as a single metric or as part of a series of informative markers. CfDNA fragment size was demonstrated as an approach for fetal fraction estimation, fetal aneuploidies, and tumor detection[95,96]. Shorter fragments have been selected in vitro or in silico in order to enrich for DNA that is more likely to be derived from a tissue of interest. Enriching for fragments from a specific tissue based on the fragment size can also enhance genetic and epigenetic signals in downstream qualitative analyses[97-99].
Beyond size profiling, the genomic location of cfDNA fragments reflects nuclear DNA architecture and gene expression in source tissues and can be used as the basis of tissue-of-origin analyses. Fragment position is known to reflect nucleosome protection of DNA from digestion. Nucleosome linker regions are more likely to be cleaved and thus play a role in preferred cfDNA fragment ends. Based on this knowledge, nucleosome occupancy maps were constructed by identifying regions with pile ups of fragment endpoints and therefore predicted to be nucleosome-free. These analyses demonstrated periodic coverage densities and nucleosome spacing that varied with chromatin state and gene activity[43,50]. Therefore, the contribution of different cell types can be ranked based on the correlation of their gene activity with the observed nucleosome spacing in gene bodies[47,100]. Different preferred end sites were found in fragments of fetal and maternal origin, and in those of malignant and non-malignant origin in liver cancer[41]. When quantifying the preferred end signature in open chromatin regions, the measure of pooled density differences of directional DNA ends could be used to trace the tissue of origin of cfDNA fragments[101]. In addition to preferred end sites, other fragmentation characteristics include end motifs and jagged ends. At the nucleotide resolution, the 5’ fragment 4-mer end motif was found to hold tissue-of-origin information as well[102]. A catalog of end motifs have altered frequencies in cancer, pregnant and transplantation subjects when compared with healthy controls and the diversity of the end motifs present in cfDNA has be used in cancer detection[102]. In cancer patients, these differences were ascribed to a deficiency in the DNASE1L3 nuclease[103,104]. Lastly, Jiang et al. explored the single-stranded overhangs by methylation analysis to infer deliberate cleavage events[105]. DNA end repair could introduce unmethylated cytosine using CpG sites or methylated cytosine using CH sites to newly synthesized ends. Interrogating the methylation levels along the DNA fragments could provide traces of enzymatic cutting of cfDNA fragments and their tissue-specific degradation patterns. The presence of such jagged or protruding ends corroborates the non-random fragments generated by endonucleases in plasma DNA and these jagged ends may be used as an indicator for relative activities of the molecular scissors, for example DNASE1 and DNASE1L3[106,107].
Clinical diagnostic applications of cfDNA
Noninvasive prenatal screening for aneuploidies
The landmark finding of cffDNA in maternal plasma paved the way for the clinical translation and commercialization of cfDNA analysis for the noninvasive prenatal screening (NIPS) of common aneuploidies. NIPS is offered to pregnant women at high risk of carrying a fetus with a common trisomy 21, 18, or 13 in many countries. In Belgium and the Netherlands, NIPS is offered as a first-tier test to all pregnant women. In a recent analysis of NIPS performance in the general obstetric population, sensitivities and specificities were 98.91% and 99.98% for trisomy 21, 97.47% and 99.99% for trisomy 18, and 100% and 99.97% for trisomy 13, respectively. The positive predictive values (PPVs) were 92.39% for trisomy 21, 84.62% for trisomy 18 and 43.95% for trisomy 13[108]. NIPS for common aneuploidies has demonstrated superior performance. In addition to the test performance. Not only does it perform well, NIPS is usually done starting from 10 weeks of pregnancy, which is earlier than or within the same time frame as invasive and first trimester combined tests. The widespread introduction of NIPS into routine prenatal care has raised concerns about disability rights, equitable access to the test, and reproductive choices[109]. Despite these ethical concerns and ongoing debate about the clinical utility, it is likely that NIPS will continue to be adopted as a first-tier screening test.
Expanded NIPS that offers additional indications for rare autosomal trisomies (RATs) and structural anomalies is available, though PPVs (ranging from 0 to 21%) for detecting RATs or structural anomalies are lower than those of the common trisomies[110]. With that, the PPVs of expanded NIPS is considerably higher than the First Trimester Combined Test. While the clinical implementation of expanded NIPS is still under debate, the prevalence of RATs and structural anomalies is as high as those of trisomy 18 and 13[111], which has motivated larger prospective studies to shed light on the clinical significance. Constitutive RATs rarely result in a viable pregnancy, however, in some cases mosaic RATs can result in adverse pregnancy outcomes, including growth impairment[112,113]. Determining the impact of these events early on in gestation could have clinical value and could support the adoption of expanded NIPS. False or inconclusive results can be the result of multiple biological factors, including confined placental mosaicism, the presence of a vanishing twin, maternal health conditions, and medication[114,115]. Continuous efforts have been made to investigate the clinical utility of the secondary findings relevant to maternal health, leading to updates in clinical management recommendations in NIPS[114].
Noninvasive prenatal screening for monogenic disorders
Noninvasive prenatal screening for monogenic diseases (NIPS-M) or noninvasive prenatal diagnosis is gaining interest in the clinic. Complimentary to NIPS for aneuploidies, NIPS-M can determine the mutational status of a fetus at high risk for a single-gene disorder using maternal plasma cfDNA analysis [Figure 5]. The test is most often offered for de novo or paternally inherited mutation diagnosis by counting the dosage of mutant and wild-type alleles or by reconstructing haplotypes. The UK National Health Service Laboratory clinically implemented NIPS-M for inherited conditions (e.g., achondroplasia, thanatophoric dysplasia and cystic fibrosis)[116,117]. A recent clinical study reported robust performance in detecting dominant monogenic diseases[78]. Though detection of recessive or maternally inherited conditions mostly remain in the research phase, a number of technologies that readily detect a range of dominant and recessive diseases have been developed[118-120]. NIPS-M has been more slowly implemented in the clinic compared to NIPS for aneuploidy, which may be due to the technical difficulty of designing bespoke tests for each family based on their genetic information.
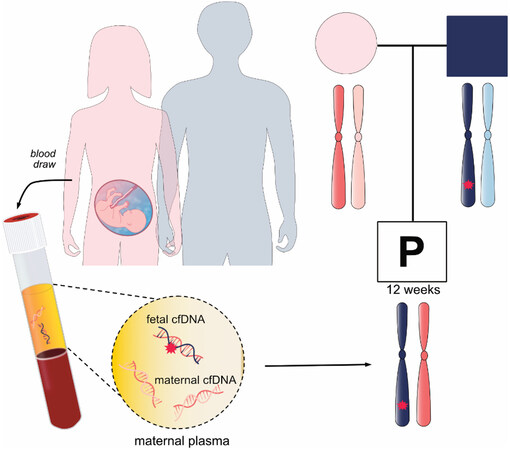
Figure 5. Noninvasive prenatal screening for monogenic disorders or noninvasive prenatal diagnosis. A family in which the father affected with an autosomal dominant disease opts for noninvasive prenatal diagnosis. Maternal plasma is taken to examine whether the fetus inherits the disease-associated allele.
Liquid biopsy in cancer
The clinical potential of liquid biopsy for cancer management has been considerably illustrated. Different ctDNA-based biomarkers or assays have been approved by the Food and Drug Administration (FDA). These include two single-cancer detection tests, namely the cobas EGFR Mutation Test v2 (Roche Molecular Diagnostics) for lung cancer and Epi proColon (Epigenomics AG) for colorectal cancer, and two targeted NGS-based pan-cancer tests: FoundationOne Liquid CDx (Foundation Medicine) and Guardant360 CDx (Guardant Health), which have been introduced as companion diagnostics to identify biomarkers in patients with advanced diseases. The noninvasive nature of cfDNA allows routine analysis of tumors that are unsafe or infeasible to biopsy [Figure 6]. Moreover, cfDNA may better reflect tumor heterogeneity harbored by distinct clonal populations. Therapeutic guidance is among the most clinically relevant current utilization of ctDNA, particularly for patients for whom standard tumor biopsies fail to yield sufficient material for analysis[121,122]. Studies have demonstrated that ctDNA testing may reveal actionable genomic mutations to guide treatment decisions[123,124]. In addition, liquid biopsy provides a way to monitor tumor burden and the development of therapy resistance in real-time. It has been shown that levels of patient-specific tumor alterations in cfDNA correlate with the overall tumor burden through serial ctDNA testing[67,125,126]. Biological resistance can be a consequence of tumor heterogeneity. ctDNA monitoring permits detection of secondary mutations associated with treatment resistance[52,127,128]. Furthermore, the longitudinal analysis of ctDNA may be extended after completion of treatment and may also serve as a means of detecting postsurgical minimal residual disease (MRD) in patients with clinically undetectable disease. Several groups have explored using postoperative ctDNA to identify patients with MRD and continued to optimize technologies[79,129,130]. A fraction of malignancies is associated with human viral infection. For the infection-induced malignancies, such as nasopharyngeal carcinoma and oropharyngeal cancer with viral DNA integrated into the host genome, plasma viral cfDNA can be quantified. Monitoring the viral cfDNA has shown prognostic potential for MRD detection[131,132]. One of the most impactful applications of ctDNA could be the potential for early cancer detection in asymptomatic populations. Early detection of cancer could increase the chances of survival. While governmental screening programs are in place for some of the cancers (e.g., breast), for many cancers, there are no screening tests available. One study evaluated analysis of Epstein-Barr virus DNA in plasma samples to screen nasopharyngeal carcinoma in asymptomatic populations, and it revealed higher positive predictive value than existing blood-based markers. A significantly higher proportion of participants with the early stage disease was identified, demonstrating the potential for early detection and early treatment intervention[133]. By far the biggest commercially driven endeavour for multi-cancer detection, the GalleriTM test, was developed by the company GRAIL[87,134]. The test was developed based on 50 different cancer types, and large-scale trials are currently ongoing to evaluate its performance for cancer screening. The combined test that uses multiparameter or multianalyte has also emerged as a key player in cancer detection[135-137]. The feasibility and safety of liquid biopsy for multi-cancer screening was largely addressed by a pioneering large clinical trial study using the DETECT-A test which combined ctDNA and protein biomarkers and performed positron emission tomography-computed tomography in patients with a positive blood test[138]. In addition to the general population, several studies have reported incidental findings of maternal malignancies during routine NIPS[139,140]. These findings have prompted large-scale or nationwide evaluations of occult maternal malignancy detection following abnormal NIPS profiles and emphasized the need to establish multidisciplinary care for clinical management of cancer in pregnancy[141,142]. Additionally, the utility of the NIPS platform - analyzing plasma cfDNA with low-pass whole-genome sequencing - for cancer detection has also been explored in high risk asymptomatic populations[74].
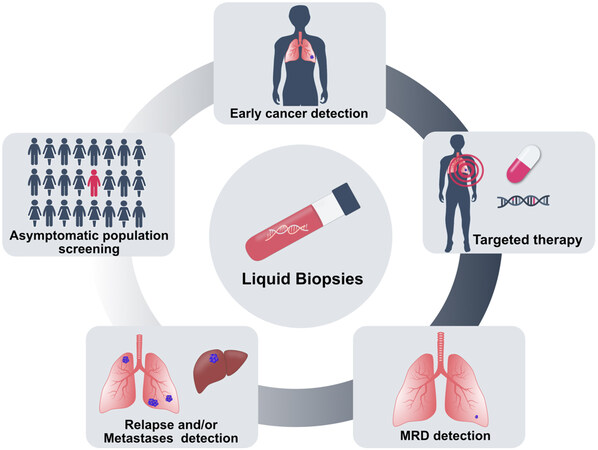
Figure 6. Clinical applications of cfDNA analysis in cancer. Analysis of cfDNA has been used to develop early cancer detection tools and ctDNA mutation analysis can guide therapy choice. After treatment, minimal residual disease (MRD) can be monitored by cfDNA analysis, and in case of resistance, cfDNA-based tests allow detection of relapse and tumor metastases. CfDNA analysis has the potential to screen cancers in the asymptomatic population.
Other applications for cfDNA analysis
The increasing interest in the use of cfDNA is not just within obstetrics and oncology. Transplantation and infectious diseases are active research areas. Based on the rationale that graft rejection entails injury and leads to increased cell death in the allograft, donor-derived cell-free DNA (dd-cfDNA) as a noninvasive marker of graft rejection has been established in solid organ and bone marrow transplants[143-145]. Quantifying the concentration of total cfDNA after transplantation, as well as the proportion of the dd-cfDNA subfraction using genotypes, allows detection of allograft injury and immunosuppression. Methylome profiling of cfDNA has also been shown to capture tissue damage in the kidney allograft[146]. Serial dd-cfDNA monitoring can provide timely views of transplant health and immunosuppressive drug effects[147].
Infection can be another major complication affecting the health of patients receiving allograft transplantation. A number of assays that concurrently monitor posttransplant rejection and infection by sequencing cfDNA have been developed[146,148,149]. Coupled with NGS, the identification of pathogen DNA in plasma of patients with sepsis, invasive fungal infection and cytomegalovirus[150-152] has broadened our ability to systematically detect clinically relevant pathogens in a noninvasive way. Viral DNA detection during NIPS has been demonstrated as well[153,154]. Blauwkamp and colleagues developed and validated a microbial cfDNA (mcfDNA) sequencing assay - the Karius test that allows detection of a wide range of infections[155]. Though this test is not able to detect RNA viruses, it has been used to identify secondary and co-infections in patients with COVID-19[156]. While tracing microbial pathogens is feasible, the analytical sensitivity could be limited by the scarcity of mcfDNA against the dominating host background[157,158]. Additionally, pathogen DNA interpretation may be complicated by reagents used for processing, sample contamination and organisms of uncertain clinical significance[158].
Virus infection can cause damage to multiple organs and tissues. Deconvoluting the cell type mixtures in cfDNA methylation profiles from COVID-19 patients, studies found elevated cfDNA levels and mapped tissue injuries particular to lung and liver compared with healthy controls or patients with other respiratory infections[159,160]. Interestingly, both studies highlighted a change in the hematopoietic cell signal as an informative marker for COVID-19 mortality prediction. While Cheng et al. observed an increase signal from erythroblasts (red blood progenitor cells) that may involve anemia and inflammation or dysregulation of erythropoiesis in COVID-19 patients[159], Andargie et al. underscored the major role of neutrophils (white blood cells) that might aggravate pulmonary inflammation and erythropoiesis as a second contributor in elevated cfDNA levels[160]. These observations may have an implication in other diseases as well. The hallmark of immune-mediated disorders is inflammation. Plasma DNA might act as an index of inflammation and damage and therefore relate to the altered genomic cfDNA seen in some patients.
“New” components of cfDNA
Along with cell-free nuclear DNA, cell-free mitochondrial DNA (cf-mtDNA) is detectable in plasma and other fluids. While cf-mtDNA research has fallen behind that of cell-free nuclear DNA, recent investigations into mitochondrial function and cf-mtDNA have elucidated its potential biological significance. Mitochondrial DNA (mtDNA) is prone to oxidative stress and may vary from dozens to thousands of copies depending on tissue origin[161]. Different forms of mitochondrial DNA, including free, particle-associated and respiratory competent mitochondria, were reported in plasma samples[162,163]. Cf-mtDNA copy number did not appear to be correlated with mtDNA copy number in cellular leukocytes, with the former being more sensitive to cellular damage and the latter reflective of cellular energetics[164]. Unlike cell-free nuclear DNA, cf-mtDNA size profile lacks nucleosome-size peaks and shows an enrichment of short fragments smaller than 100 bp[9] due to the lack of histones. Though limited by different pre-analytical protocols and mixed conclusions, cf-mtDNA has been found to be elevated in multiple pathologies, including trauma, sepsis, neurological disorders, and cancer[164-168]. A highlight of recent cf-mtDNA research comes from the identification of both intact circular and fragmented linear cf-mtDNA in plasma DNA. Of particular interest, the authors observed that non-hematopoietically derived cf-mtDNA was predominantly in a linear form, whereas hematopoietically derived cf-mtDNA was primarily circular using a transplantation model[169]. It is noteworthy that analogous observations regarding fetal and maternal cf-mtDNA in surrogate pregnancies have subsequently been reported by the group[170]. The fetal-derived cf-mtDNA mainly existed in a linear form, but about half of maternal-derived cf-mtDNA appeared to be in a circular form. Although there is a major knowledge gap between the triggers that connect pathophysiological states to the cf-mtDNA release into circulation and different biological forms of cf-mtDNA, these findings might prompt the development of cf-mtDNA assays.
Another circular DNA - extrachromosomal circular DNA (eccDNA) has been known for a long time. The recent development of an assay to quantify eccDNA in plasma DNA (referred to as cf-eccDNA throughout the text) entices renewed interest. This circular DNA exists as small (200 to 400 bp), non-amplified eccDNA in normal cells[171] and as large (1.29 Mb EGFRvIII were detected), copy number-amplified extrachromosomal circular DNA (referred to as ecDNA to differentiate from eccDNA) that is primarily found in cancer cells[172]. Recent studies demonstrated that ecDNA is common in human cancer cells (up to hundreds of ecDNA molecules per cell), with the highest abundance in brain tumors. Support for the presence of cf-eccDNA in circulation was first shown in two studies that enriched circular DNA by exonuclease digestion of the background linear DNA[173,174]. These studies demonstrated that cf-eccDNA harbors characteristics of eccDNA that tends to be generated from regions with high GC content, gene density, active chromatin marks and a high frequency of direct repeats flanking eccDNA junctions. Furthermore, the size of cf-eccDNA tended to be longer when comparing pre- to post-surgery in lung and ovarian cancer patients, possibly implying that cancer-associated eccDNAs are longer than the normal counterparts[173]. It has been found that fetal-derived eccDNA is present in the plasma of pregnant women. The cf-eccDNA fragment size distribution was shown to have a small peak at 202 bp, another major peak at 338 bp and a 10 bp periodicity in the vicinity of this peak. The fetal cf-eccDNA tended to be shorter than the maternal counterparts, consistent with the phenomenon in linear nuclear cfDNA[175]. Reminiscent of nuclear cfDNA patterns, fetal cf-eccDNA shows relatively lower methylation levels than those of the maternal cf-eccDNA and clears rapidly after delivery with a half-life of 30 min. Longer cf-eccDNA fragments appears to carry higher methylation densities in plasma of both pregnant and non-pregnant subjects[176]. Furthermore, nuclease activity on cf-eccDNA was reported, showing that DNASE1L3 affected eccDNA characteristics[177].
New frontiers in cfDNA biology
CfDNA has long been considered as cell death debris. However, growing evidence points to potential functional aspects of cfDNA, including its role in horizontal genetic transfer, cellular signaling, oxidative stress response, and innate immunity[1,178,179]. Immunological properties of cfDNA can be particularly relevant to certain disease pathologies. Interplaying with nuclease biology, the presence of endogenous DNase allows proper DNA degradation and prevents inflammatory stimulation, whereas DNase-deficient mice were prone to develop autoimmune pathologies[180] and exhibited cfDNA fragmentation aberrations[103]. Indeed, DNase1 activity was found to be substantially lower in patients with systemic lupus erythematosus (SLE)[181]. In pathological conditions like lupus, cfDNA complexed with other proteins may act as proinflammatory stimulants of toll-like receptors (TLR) that elicit the inflammatory response[107]. Such mechanisms of recognizing extracellular DNA as damage-associated by the innate immune system has been shown in the case of cf-mtDNA in patients with trauma[165]. Triggered by the innate immune response, NETosis that initiates the formation of Neutrophil Extracellular Traps (NETs) could promote thrombosis and mediate tissue damage[182]. Formed as extracellular web-like structure with antimicrobial proteins, the NETs have been suggested to play a role in the pathogenesis of multiple diseases, including lupus, sepsis and tumor progression[183-186]. Remarkably, studies have found that host DNase1 and DNase1L3 act as dual protection systems to degrade NETs and in the absence of DNase1 and DNase1L3, intravascular NETs form clots and result in organ damage[187]. The aforementioned studies that map tissue injury in COVID-19 patients may imply potential immunological effects in cfDNA. Additional pertinent evidence reported strong correlation between levels of cfDNA and traditional inflammatory markers, in particular, absolute neutrophil count and myeloperoxidase-DNA that is regarded as a specific marker of NETs in hospitalized COVID-19 patients[188]. In transplantation, elevated dd-cfDNA in the presence of allograft injury may indicate its active role in immunological processes as well[189]. Rather than showing immunostimulatory characteristics itself, cfDNA may be a non-specific (pro)inflammation and (pro)coagulation marker in numerous inflammatory and cell death pathways. The question is to what extent the immunological effects are reflected in cfDNA and how the changes can be effectively captured.
Concluding remarks
Fragmented DNA constantly enters our circulation following passive and active release mechanisms. The composition of tissues contributing to cfDNA differs from one disease to another, and across different physiological states. With the rapid maturity of next generation sequencing and bioinformatics analysis, cfDNA analysis has led to robust copy number detection tools in prenatal testing and tumor liquid biopsy. Yet clinical applications lag behind our expanding understanding of cfDNA biology. In particular, epigenetic and fragmentomic characteristics of cfDNA are an untapped source of potential clinical markers in a wide range of diseases. Novel experimental and computational methods capable of simultaneously capturing and integrating cfDNA modalities present attractive opportunities to push analyses further and may allow for synergistic effects.
DECLARATIONS
Authors’ contributionsWrote the manuscript: Che H, Stanley K
All authors revised and reviewed the manuscript.
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was supported by Research Foundation-Flanders (FWO) G080217N to JRV, FWO-SBO-MICADO (S003422N) and KU Leuven funding (no. C1/018 to JRV).
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2022.
REFERENCES
1. Thierry AR, El Messaoudi S, Gahan PB, Anker P, Stroun M. Origins, structures, and functions of circulating DNA in oncology. Cancer Metastasis Rev 2016;35:347-76.
2. Bronkhorst AJ, Ungerer V, Holdenrieder S. The emerging role of cell-free DNA as a molecular marker for cancer management. Biomol Detect Quantif 2019;17:100087.
3. Lui YY, Chik K, Chiu RW, Ho C, Lam CW, Lo YD. Predominant hematopoietic origin of cell-free DNA in plasma and serum after sex-mismatched bone marrow transplantation. Clin Chem 2002;48:421-7.
4. Sun K, Jiang P, Chan KC, et al. Plasma DNA tissue mapping by genome-wide methylation sequencing for noninvasive prenatal, cancer, and transplantation assessments. Proc Natl Acad Sci USA 2015;112:E5503-12.
6. Moss J, Magenheim J, Neiman D, et al. Comprehensive human cell-type methylation atlas reveals origins of circulating cell-free DNA in health and disease. Nat Commun 2018;9:5068.
7. Giacona MB, Ruben GC, Iczkowski KA, Roos TB, Porter DM, Sorenson GD. Cell-free DNA in human blood plasma: length measurements in patients with pancreatic cancer and healthy controls. Pancreas 1998;17:89-97.
8. Jiang P, Lo YMD. The long and short of circulating cell-free DNA and the ins and outs of molecular diagnostics. Trends Genet 2016;32:360-71.
9. Lo YM, Chan KC, Sun H, et al. Maternal plasma DNA sequencing reveals the genome-wide genetic and mutational profile of the fetus. Sci Transl Med 2010;2:61ra91.
10. Jahr S, Hentze H, Englisch S, et al. DNA fragments in the blood plasma of cancer patients: quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res 2001;61:1659-1665.
11. Fan HC, Blumenfeld YJ, Chitkara U, Hudgins L, Quake SR. Analysis of the size distributions of fetal and maternal cell-free DNA by paired-end sequencing. Clin Chem 2010;56:1279-86.
12. Ivanov M, Baranova A, Butler T, Spellman P, Mileyko V. Non-random fragmentation patterns in circulating cell-free DNA reflect epigenetic regulation. BMC Genomics 2015;16 Suppl 13:S1.
13. Sanchez C, Roch B, Mazard T, et al. Circulating nuclear DNA structural features, origins, and complete size profile revealed by fragmentomics. JCI Insight 2021;6:144561.
14. Aucamp J, Bronkhorst AJ, Badenhorst CPS, Pretorius PJ. The diverse origins of circulating cell-free DNA in the human body: a critical re-evaluation of the literature. Biol Rev Camb Philos Soc 2018;93:1649-83.
15. Kustanovich A, Schwartz R, Peretz T, Grinshpun A. Life and death of circulating cell-free DNA. Cancer Biol Ther 2019;20:1057-67.
16. Elkon KB. Review: cell death, nucleic acids, and immunity: inflammation beyond the grave. Arthritis Rheumatol 2018;70:805-16.
17. Stroun M, Lyautey J, Lederrey C, Olson-sand A, Anker P. About the possible origin and mechanism of circulating DNA. Clinica Chimica Acta 2001;313:139-42.
18. Lau TW, Leung TN, Chan LYS, et al. Fetal DNA clearance from maternal plasma is impaired in preeclampsia. Clin Chem 2002;48:2141-2146.
19. Courtney PA, Crockard AD, Williamson K, Irvine AE, Kennedy RJ, Bell AL. Increased apoptotic peripheral blood neutrophils in systemic lupus erythematosus: relations with disease activity, antibodies to double stranded DNA, and neutropenia. Ann Rheum Dis 1999;58:309-14.
20. Heitzer E, Auinger L, Speicher MR. Cell-free DNA and apoptosis: how dead cells inform about the living. Trends Mol Med 2020;26:519-28.
21. Han DSC, Ni M, Chan RWY, et al. The biology of cell-free DNA fragmentation and the roles of DNASE1, DNASE1L3, and DFFB. Am J Hum Genet 2020;106:202-14.
22. Duvvuri B, Lood C. Cell-free DNA as a biomarker in autoimmune rheumatic diseases. Front Immunol 2019;10:502.
23. Al-Mayouf SM, Sunker A, Abdwani R, et al. Loss-of-function variant in DNASE1L3 causes a familial form of systemic lupus erythematosus. Nat Genet 2011;43:1186-8.
24. Leffler J, Ciacma K, Gullstrand B, Bengtsson AA, Martin M, Blom AM. A subset of patients with systemic lupus erythematosus fails to degrade DNA from multiple clinically relevant sources. Arthritis Res Ther 2015;17:205.
25. Diehl F, Schmidt K, Choti MA, et al. Circulating mutant DNA to assess tumor dynamics. Nat Med 2008;14:985-90.
26. Lo YMD, Corbetta N, Chamberlain PF, et al. Presence of fetal DNA in maternal plasma and serum. The Lancet 1997;350:485-7.
27. Alberry M, Maddocks D, Jones M, et al. Free fetal DNA in maternal plasma in anembryonic pregnancies: confirmation that the origin is the trophoblast. Prenat Diagn 2007;27:415-8.
28. Karakas B, Qubbaj W, Al-Hassan S, Coskun S. Noninvasive digital detection of fetal DNA in plasma of 4-week-pregnant women following in vitro fertilization and embryo transfer. PLoS One 2015;10:e0126501.
29. Chim SS, Tong YK, Chiu RW, et al. Detection of the placental epigenetic signature of the maspin gene in maternal plasma. Proc Natl Acad Sci USA 2005;102:14753-8.
30. Chiu RW, Chim SS, Wong IH, et al. Hypermethylation of RASSF1A in human and rhesus placentas. Am J Pathol 2007;170:941-50.
31. Lun FM, Chiu RW, Sun K, et al. Noninvasive prenatal methylomic analysis by genomewide bisulfite sequencing of maternal plasma DNA. Clin Chem 2013;59:1583-94.
32. Masuzaki H, Miura K, Yoshiura KI, Yoshimura S, Niikawa N, Ishimaru T. Detection of cell free placental DNA in maternal plasma: direct evidence from three cases of confined placental mosaicism. J Med Genet 2004;41:289-92.
33. Faas BH, de Ligt J, Janssen I, et al. Non-invasive prenatal diagnosis of fetal aneuploidies using massively parallel sequencing-by-ligation and evidence that cell-free fetal DNA in the maternal plasma originates from cytotrophoblastic cells. Expert Opin Biol Ther 2012;12 Suppl 1:S19-26.
34. Villela D, Che H, Van Ghelue M, et al. Fetal sex determination in twin pregnancies using non-invasive prenatal testing. NPJ Genom Med 2019;4:15.
35. Levine RJ, Qian C, Leshane ES, et al. Two-stage elevation of cell-free fetal DNA in maternal sera before onset of preeclampsia. Am J Obstet Gynecol 2004;190:707-13.
36. Hahn S, Rusterholz C, Hösli I, Lapaire O. Cell-free nucleic acids as potential markers for preeclampsia. Placenta 2011;32 Suppl:S17-20.
37. Karapetian АО, Baev ОR, Sadekova АА, Krasnyi АМ, Sukhikh GT. Cell-free foetal DNA as a useful marker for preeclampsia prediction. Reprod Sci 2021;28:1563-9.
38. Palomaki GE, Kloza EM, Lambert-Messerlian GM, et al. DNA sequencing of maternal plasma to detect Down syndrome: an international clinical validation study. Genet Med 2011;13:913-20.
39. Chiu RWK, Lo YMD. Cell-free fetal DNA coming in all sizes and shapes. Prenat Diagn 2021;41:1193-201.
40. Yu SC, Lee SW, Jiang P, et al. High-resolution profiling of fetal DNA clearance from maternal plasma by massively parallel sequencing. Clin Chem 2013;59:1228-37.
41. Sun K, Jiang P, Wong AIC, et al. Size-tagged preferred ends in maternal plasma DNA shed light on the production mechanism and show utility in noninvasive prenatal testing. Proc Natl Acad Sci USA 2018;115:E5106-14.
42. Kim SK, Hannum G, Geis J, et al. Determination of fetal DNA fraction from the plasma of pregnant women using sequence read counts. Prenat Diagn 2015;35:810-5.
43. Straver R, Oudejans CB, Sistermans EA, Reinders MJ. Calculating the fetal fraction for noninvasive prenatal testing based on genome-wide nucleosome profiles. Prenat Diagn 2016;36:614-21.
44. Stroun M, Anker P, Maurice P, Lyautey J, Lederrey C, Beljanski M. Neoplastic characteristics of the DNA found in the plasma of cancer patients. Oncology 1989;46:318-22.
45. Chen XQ, Stroun M, Magnenat JL, et al. Microsatellite alterations in plasma DNA of small cell lung cancer patients. Nat Med 1996;2:1033-5.
46. Nawroz H, Koch W, Anker P, Stroun M, Sidransky D. Microsatellite alterations in serum DNA of head and neck cancer patients. Nat Med 1996;2:1035-7.
47. Ulz P, Thallinger GG, Auer M, et al. Inferring expressed genes by whole-genome sequencing of plasma DNA. Nature Genetics. 2016;48:1273.
48. De Mattos-Arruda L, Weigelt B, Cortes J, et al. Capturing intra-tumor genetic heterogeneity by de novo mutation profiling of circulating cell-free tumor DNA: a proof-of-principle. Ann Oncol 2014;25:1729-35.
49. Murtaza M, Dawson SJ, Pogrebniak K, et al. Multifocal clonal evolution characterized using circulating tumour DNA in a case of metastatic breast cancer. Nat Commun 2015;6:8760.
50. Snyder MW, Kircher M, Hill AJ, Daza RM, Shendure J. Cell-free DNA comprises an in vivo nucleosome footprint that informs its tissues-of-origin. Cell 2016;164:57-68.
51. Mouliere F, Thierry AR. The importance of examining the proportion of circulating DNA originating from tumor, microenvironment and normal cells in colorectal cancer patients. Expert Opin Biol Ther 2012;12 Suppl 1:S209-15.
52. Bettegowda C, Sausen M, Leary RJ, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med 2014;6:224ra24.
53. Tie J, Kinde I, Wang Y, et al. Circulating tumor DNA as an early marker of therapeutic response in patients with metastatic colorectal cancer. Ann Oncol 2015;26:1715-22.
54. Gray ES, Rizos H, Reid AL, et al. Circulating tumor DNA to monitor treatment response and detect acquired resistance in patients with metastatic melanoma. Oncotarget 2015;6:42008-18.
55. Vanderstichele A, Busschaert P, Smeets D, et al. Chromosomal instability in cell-free DNA as a highly specific biomarker for detection of ovarian cancer in women with adnexal masses. Clin Cancer Res 2017;23:2223-31.
56. Lenaerts L, Che H, Brison N, et al. Breast cancer detection and treatment monitoring using a noninvasive prenatal testing platform: utility in pregnant and nonpregnant populations. Clin Chem 2020;66:1414-23.
57. Smith CG, Moser T, Mouliere F, et al. Comprehensive characterization of cell-free tumor DNA in plasma and urine of patients with renal tumors. Genome Med 2020;12:23.
58. Markus H, Zhao J, Contente-Cuomo T, et al. Analysis of recurrently protected genomic regions in cell-free DNA found in urine. Sci Transl Med 2021;13:eaaz3088.
59. Mouliere F, Robert B, Arnau Peyrotte E, et al. High fragmentation characterizes tumour-derived circulating DNA. PLoS One 2011;6:e23418.
60. Kerachian MA, Azghandi M, Mozaffari-Jovin S, Thierry AR. Guidelines for pre-analytical conditions for assessing the methylation of circulating cell-free DNA. Clin Epigenetics 2021;13:193.
61. van der Pol Y, Moldovan N, Verkuijlen S, et al. The effect of preanalytical and physiological variables on cell-free DNA fragmentation. Clin Chem 2022;68:803-13.
62. Van Paemel R, De Koker A, Caggiano C, et al. Genome-wide study of the effect of blood collection tubes on the cell-free DNA methylome. Epigenetics 2021;16:797-807.
63. Kwak DW, Kim SY, Kim HJ, Lim JH, Kim YH, Ryu HM. Maternal total cell-free DNA in preeclampsia with and without intrauterine growth restriction. Sci Rep 2020;10:11848.
64. Tug S, Helmig S, Menke J, et al. Correlation between cell free DNA levels and medical evaluation of disease progression in systemic lupus erythematosus patients. Cell Immunol 2014;292:32-9.
65. Chen E, Cario CL, Leong L, et al. Cell-free DNA concentration and fragment size as a biomarker for prostate cancer. Sci Rep 2021;11:5040.
66. Sifakis S, Koukou Z, Spandidos DA. Cell-free fetal DNA and pregnancy-related complications (review). Mol Med Rep 2015;11:2367-72.
67. Forshew T, Murtaza M, Parkinson C, et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci Transl Med 2012;4:136ra68.
68. Chiu RW, Chan KC, Gao Y, et al. Noninvasive prenatal diagnosis of fetal chromosomal aneuploidy by massively parallel genomic sequencing of DNA in maternal plasma. Proc Natl Acad Sci USA 2008;105:20458-63.
69. Fan HC, Blumenfeld YJ, Chitkara U, Hudgins L, Quake SR. Noninvasive diagnosis of fetal aneuploidy by shotgun sequencing DNA from maternal blood. Proc Natl Acad Sci USA 2008;105:16266-71.
70. Ehrich M, Deciu C, Zwiefelhofer T, et al. Noninvasive detection of fetal trisomy 21 by sequencing of DNA in maternal blood: a study in a clinical setting. Am J Obstet Gynecol 2011;204:205.e1-11.
71. Bianchi DW, Platt LD, Goldberg JD, Abuhamad AZ, Sehnert AJ, Rava RP. MatErnal BLood IS Source to Accurately diagnose fetal aneuploidy (MELISSA) Study Group. Genome-wide fetal aneuploidy detection by maternal plasma DNA sequencing. Obstet Gynecol 2012;119:890-901.
72. Chiu RW, Akolekar R, Zheng YW, et al. Non-invasive prenatal assessment of trisomy 21 by multiplexed maternal plasma DNA sequencing: large scale validity study. BMJ 2011;342:c7401.
73. Heitzer E, Ulz P, Belic J, et al. Tumor-associated copy number changes in the circulation of patients with prostate cancer identified through whole-genome sequencing. Genome Med 2013;5:30.
74. Lenaerts L, Vandenberghe P, Brison N, et al. Genomewide copy number alteration screening of circulating plasma DNA: potential for the detection of incipient tumors. Ann Oncol 2019;30:85-95.
75. Newman AM, Bratman SV, To J, et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med 2014;20:548-54.
76. Newman AM, Lovejoy AF, Klass DM, et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat Biotechnol 2016;34:547-55.
77. Wan JCM, Heider K, Gale D, et al. ctDNA monitoring using patient-specific sequencing and integration of variant reads. Sci Transl Med 2020;12:eaaz8084.
78. Zhang J, Li J, Saucier JB, et al. Non-invasive prenatal sequencing for multiple Mendelian monogenic disorders using circulating cell-free fetal DNA. Nat Med 2019;25:439-47.
79. Kurtz DM, Soo J, Co Ting Keh L, et al. Enhanced detection of minimal residual disease by targeted sequencing of phased variants in circulating tumor DNA. Nat Biotechnol 2021;39:1537-47.
80. Leary RJ, Kinde I, Diehl F, et al. Development of personalized tumor biomarkers using massively parallel sequencing. Sci Transl Med 2010;2:20ra14.
81. Clark TA, Chung JH, Kennedy M, et al. Analytical validation of a hybrid capture-based next-generation sequencing clinical assay for genomic profiling of cell-free circulating tumor DNA. J Mol Diagn 2018;20:686-702.
82. Wong IHN, Lo YMD, Zhang J, et al. Detection of aberrant p16 methylation in the plasma and serum of liver cancer patients. Cancer Res 1999;59:71-73.
83. Poon LL, Leung TN, Lau TK, Chow KC, Lo YD. Differential DNA methylation between fetus and mother as a strategy for detecting fetal DNA in maternal plasma. Clin Chem 2002;48:35-41.
84. Chan KC, Ding C, Gerovassili A, et al. Hypermethylated RASSF1A in maternal plasma: a universal fetal DNA marker that improves the reliability of noninvasive prenatal diagnosis. Clin Chem 2006;52:2211-8.
85. Chan KC, Jiang P, Chan CW, et al. Noninvasive detection of cancer-associated genome-wide hypomethylation and copy number aberrations by plasma DNA bisulfite sequencing. Proc Natl Acad Sci USA 2013;110:18761-8.
86. Jensen TJ, Kim SK, Zhu Z, et al. Whole genome bisulfite sequencing of cell-free DNA and its cellular contributors uncovers placenta hypomethylated domains. Genome Biol 2015;16:78.
87. Liu MC, Oxnard GR, Klein EA, Swanton C, Seiden MV. CCGA Consortium. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann Oncol 2020;31:745-59.
88. Guo S, Diep D, Plongthongkum N, Fung HL, Zhang K, Zhang K. Identification of methylation haplotype blocks aids in deconvolution of heterogeneous tissue samples and tumor tissue-of-origin mapping from plasma DNA. Nat Genet 2017;49:635-42.
89. Shen SY, Singhania R, Fehringer G, et al. Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nature 2018;563:579-83.
90. Erger F, Nörling D, Borchert D, et al. cfNOMe - a single assay for comprehensive epigenetic analyses of cell-free DNA. Genome Med 2020;12:54.
91. Vaisvila R, Ponnaluri VKC, Sun Z, et al. Enzymatic methyl sequencing detects DNA methylation at single-base resolution from picograms of DNA. Genome Res 2021;31:1280-9.
92. Liu Y, Siejka-Zielińska P, Velikova G, et al. Bisulfite-free direct detection of 5-methylcytosine and 5-hydroxymethylcytosine at base resolution. Nat Biotechnol 2019;37:424-9.
93. Yu SCY, Jiang P, Peng W, et al. Single-molecule sequencing reveals a large population of long cell-free DNA molecules in maternal plasma. Proc Natl Acad Sci USA 2021;118:e2114937118.
94. Sadeh R, Sharkia I, Fialkoff G, et al. ChIP-seq of plasma cell-free nucleosomes identifies gene expression programs of the cells of origin. Nat Biotechnol 2021;39:586-98.
95. Yu SC, Chan KC, Zheng YW, et al. Size-based molecular diagnostics using plasma DNA for noninvasive prenatal testing. Proc Natl Acad Sci USA 2014;111:8583-8.
96. Cristiano S, Leal A, Phallen J, et al. Genome-wide cell-free DNA fragmentation in patients with cancer. Nature 2019;570:385-9.
97. Mouliere F, Chandrananda D, Piskorz AM, et al. Enhanced detection of circulating tumor DNA by fragment size analysis. Sci Transl Med 2018;10:eaat4921.
98. Budis J, Gazdarica J, Radvanszky J, et al. Combining count- and length-based z-scores leads to improved predictions in non-invasive prenatal testing. Bioinformatics 2019;35:1284-91.
99. Liu J, Zhao H, Huang Y, et al. Genome-wide cell-free DNA methylation analyses improve accuracy of non-invasive diagnostic imaging for early-stage breast cancer. Mol Cancer 2021;20:36.
100. Ulz P, Perakis S, Zhou Q, et al. Inference of transcription factor binding from cell-free DNA enables tumor subtype prediction and early detection. Nat Commun 2019;10:4666.
101. Sun K, Jiang P, Cheng SH, et al. Orientation-aware plasma cell-free DNA fragmentation analysis in open chromatin regions informs tissue of origin. Genome Res 2019;29:418-27.
102. Jiang P, Sun K, Peng W, et al. Plasma DNA end-motif profiling as a fragmentomic marker in cancer, pregnancy, and transplantation. Cancer Discov 2020;10:664-73.
103. Serpas L, Chan RWY, Jiang P, et al. Dnase1l3 deletion causes aberrations in length and end-motif frequencies in plasma DNA. Proc Natl Acad Sci USA 2019;116:641-9.
104. Chan RWY, Serpas L, Ni M, et al. Plasma DNA profile associated with DNASE1L3 gene mutations: clinical observations, relationships to nuclease substrate preference, and in vivo correction. Am J Hum Genet 2020;107:882-94.
105. Jiang P, Xie T, Ding SC, et al. Detection and characterization of jagged ends of double-stranded DNA in plasma. Genome Res 2020;30:1144-53.
106. Suzuki N, Kamataki A, Yamaki J, Homma Y. Characterization of circulating DNA in healthy human plasma. Clin Chim Acta 2008;387:55-8.
108. Van Den Bogaert K, Lannoo L, Brison N, et al. Outcome of publicly funded nationwide first-tier noninvasive prenatal screening. Genet Med 2021;23:1137-42.
109. Ravitsky V, Roy MC, Haidar H, et al. The emergence and global spread of noninvasive prenatal testing. Annu Rev Genomics Hum Genet 2021;22:309-38.
110. Petersen AK, Cheung SW, Smith JL, et al. Positive predictive value estimates for cell-free noninvasive prenatal screening from data of a large referral genetic diagnostic laboratory. Am J Obstet Gynecol 2017;217:691.e1-6.
111. Wellesley D, Dolk H, Boyd PA, et al. Rare chromosome abnormalities, prevalence and prenatal diagnosis rates from population-based congenital anomaly registers in Europe. Eur J Hum Genet 2012;20:521-6.
112. Pertile MD, Halks-Miller M, Flowers N, et al. Rare autosomal trisomies, revealed by maternal plasma DNA sequencing, suggest increased risk of feto-placental disease. Sci Transl Med 2017;9:eaan1240.
113. Benn P, Malvestiti F, Grimi B, Maggi F, Simoni G, Grati FR. Rare autosomal trisomies: comparison of detection through cell-free DNA analysis and direct chromosome preparation of chorionic villus samples. Ultrasound Obstet Gynecol 2019;54:458-67.
114. Bianchi DW, Chiu RWK. Sequencing of circulating cell-free DNA during pregnancy. N Engl J Med 2018;379:464-73.
115. Bianchi DW. Cherchez la femme: maternal incidental findings can explain discordant prenatal cell-free DNA sequencing results. Genet Med 2018;20:910-7.
116. Chitty LS, Mason S, Barrett AN, et al. Non-invasive prenatal diagnosis of achondroplasia and thanatophoric dysplasia: next-generation sequencing allows for a safer, more accurate, and comprehensive approach. Prenat Diagn 2015;35:656-62.
117. Lench N, Barrett A, Fielding S, et al. The clinical implementation of non-invasive prenatal diagnosis for single-gene disorders: challenges and progress made. Prenat Diagn 2013;33:555-62.
118. Chiu EKL, Hui WWI, Chiu RWK. cfDNA screening and diagnosis of monogenic disorders - where are we heading? Prenat Diagn 2018;38:52-8.
119. Scotchman E, Shaw J, Paternoster B, Chandler N, Chitty LS. Non-invasive prenatal diagnosis and screening for monogenic disorders. Eur J Obstet Gynecol Reprod Biol 2020;253:320-7.
120. Che H, Villela D, Dimitriadou E, et al. Noninvasive prenatal diagnosis by genome-wide haplotyping of cell-free plasma DNA. Genet Med 2020;22:962-73.
121. Zill OA, Greene C, Sebisanovic D, et al. Cell-Free DNA next-generation sequencing in pancreatobiliary carcinomas. Cancer Discov 2015;5:1040-8.
122. Oxnard GR, Thress KS, Alden RS, et al. Association between plasma genotyping and outcomes of treatment with osimertinib (AZD9291) in advanced non-small-cell lung cancer. J Clin Oncol 2016;34:3375-82.
123. Shu Y, Wu X, Tong X, et al. Circulating tumor DNA mutation profiling by targeted next generation sequencing provides guidance for personalized treatments in multiple cancer types. Sci Rep 2017;7:583.
124. Adalsteinsson VA, Ha G, Freeman SS, et al. Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nat Commun 2017;8:1324.
125. Dawson SJ, Tsui DW, Murtaza M, et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med 2013;368:1199-209.
126. Siravegna G, Mussolin B, Buscarino M, et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat Med 2015;21:795-801.
127. Chabon JJ, Simmons AD, Lovejoy AF, et al. Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients. Nat Commun 2016;7:11815.
128. Corcoran RB, Chabner BA. Application of cell-free DNA analysis to cancer treatment. N Engl J Med 2018;379:1754-65.
129. Chaudhuri AA, Chabon JJ, Lovejoy AF, et al. Early detection of molecular residual disease in localized lung cancer by circulating tumor DNA profiling. Cancer Discov 2017;7:1394-403.
130. McDonald BR, Contente-Cuomo T, Sammut SJ, et al. Personalized circulating tumor DNA analysis to detect residual disease after neoadjuvant therapy in breast cancer. Sci Transl Med 2019;11:eaax7392.
131. Chan DCT, Lam WKJ, Hui EP, et al. Improved risk stratification of nasopharyngeal cancer by targeted sequencing of Epstein-Barr virus DNA in post-treatment plasma. Ann Oncol 2022;33:794-803.
132. Hanna GJ, Supplee JG, Kuang Y, et al. Plasma HPV cell-free DNA monitoring in advanced HPV-associated oropharyngeal cancer. Ann Oncol 2018;29:1980-6.
133. Chan KCA, Woo JKS, King A, et al. Analysis of plasma epstein-barr virus DNA to screen for nasopharyngeal cancer. N Engl J Med 2017;377:513-22.
134. Klein EA, Richards D, Cohn A, et al. Clinical validation of a targeted methylation-based multi-cancer early detection test using an independent validation set. Ann Oncol 2021;32:1167-77.
135. Cohen JD, Li L, Wang Y, et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 2018;359:926-30.
136. Sefrioui D, Blanchard F, Toure E, et al. Diagnostic value of CA19.9, circulating tumour DNA and circulating tumour cells in patients with solid pancreatic tumours. Br J Cancer 2017;117:1017-25.
137. Cohen JD, Javed AA, Thoburn C, et al. Combined circulating tumor DNA and protein biomarker-based liquid biopsy for the earlier detection of pancreatic cancers. Proc Natl Acad Sci USA 2017;114:10202-7.
138. Lennon AM, Buchanan AH, Kinde I, et al. Feasibility of blood testing combined with PET-CT to screen for cancer and guide intervention. Science 2020;369:eabb9601.
139. Vandenberghe P, Wlodarska I, Tousseyn T, et al. Non-invasive detection of genomic imbalances in Hodgkin/Reed-Sternberg cells in early and advanced stage Hodgkin's lymphoma by sequencing of circulating cell-free DNA: a technical proof-of-principle study. Lancet Haematol 2015;2:e55-65.
140. Amant F, Verheecke M, Wlodarska I, et al. Presymptomatic identification of cancers in pregnant women during noninvasive prenatal testing. JAMA Oncol 2015;1:814-9.
141. Lenaerts L, Brison N, Maggen C, et al. Comprehensive genome-wide analysis of routine non-invasive test data allows cancer prediction: a single-center retrospective analysis of over 85,000 pregnancies. EClinicalMedicine 2021;35:100856.
142. Heesterbeek CJ, Aukema SM, Galjaard RH, et al. Dutch NIPT consortium. noninvasive prenatal test results indicative of maternal malignancies: a nationwide genetic and clinical follow-up study. J Clin Oncol 2022;40:2426-35.
143. Snyder TM, Khush KK, Valantine HA, Quake SR. Universal noninvasive detection of solid organ transplant rejection. Proc Natl Acad Sci USA 2011;108:6229-34.
144. De Vlaminck I, Valantine HA, Snyder TM, et al. Circulating cell-free DNA enables noninvasive diagnosis of heart transplant rejection. Sci Transl Med 2014;6:241ra77.
145. Sharon E, Shi H, Kharbanda S, et al. Quantification of transplant-derived circulating cell-free DNA in absence of a donor genotype. PLoS Comput Biol 2017;13:e1005629.
146. Cheng AP, Burnham P, Lee JR, et al. A cell-free DNA metagenomic sequencing assay that integrates the host injury response to infection. Proc Natl Acad Sci USA 2019;116:18738-44.
147. Schütz E, Asendorf T, Beck J, et al. Time-dependent apparent increase in dd-cfDNA percentage in clinically stable patients between one and five years following kidney transplantation. Clin Chem 2020;66:1290-9.
148. Zhou Y, Yang G, Liu H, et al. A Noninvasive and donor-independent method simultaneously monitors rejection and infection in patients with organ transplant. Transplant Proc 2019;51:1699-705.
149. Cheng AP, Cheng MP, Loy CJ, et al. Cell-free DNA profiling informs all major complications of hematopoietic cell transplantation. Proc Natl Acad Sci USA 2022;119:e2113476118.
150. Long Y, Zhang Y, Gong Y, et al. Diagnosis of sepsis with cell-free DNA by next-generation sequencing technology in ICU patients. Arch Med Res 2016;47:365-71.
151. Hong DK, Blauwkamp TA, Kertesz M, Bercovici S, Truong C, Banaei N. Liquid biopsy for infectious diseases: sequencing of cell-free plasma to detect pathogen DNA in patients with invasive fungal disease. Diagn Microbiol Infect Dis 2018;92:210-3.
152. Chesnais V, Ott A, Chaplais E, et al. Using massively parallel shotgun sequencing of maternal plasmatic cell-free DNA for cytomegalovirus DNA detection during pregnancy: a proof of concept study. Sci Rep 2018;8:4321.
153. Liu S, Huang S, Chen F, et al. Genomic analyses from non-invasive prenatal testing reveal genetic associations, patterns of viral infections, and Chinese population history. Cell 2018;175:347-359.e14.
154. Linthorst J, Baksi MMM, Welkers MRA, Sistermans EA. The cell-free DNA virome of 108,349 Dutch pregnant women. Prenat Diagn 2022; doi: 10.1002/pd.6143.
155. Blauwkamp TA, Thair S, Rosen MJ, et al. Analytical and clinical validation of a microbial cell-free DNA sequencing test for infectious disease. Nat Microbiol 2019;4:663-74.
156. Kitsios GD, Bain W, Al-Yousif N, et al. Plasma microbial cell-free DNA load is associated with mortality in patients with COVID-19. Respir Res 2021;22:24.
157. Benamu E, Gajurel K, Anderson JN, et al. Plasma microbial cell-free DNA next-generation sequencing in the diagnosis and management of febrile neutropenia. Clin Infect Dis 2022;74:1659-68.
158. Gu W, Miller S, Chiu CY. Clinical metagenomic next-generation sequencing for pathogen detection. Annu Rev Pathol 2019;14:319-38.
159. Cheng AP, Cheng MP, Gu W, et al. Cell-free DNA tissues of origin by methylation profiling reveals significant cell, tissue, and organ-specific injury related to COVID-19 severity. Med 2021;2:411-422.e5.
160. Andargie TE, Tsuji N, Seifuddin F, et al. Cell-free DNA maps COVID-19 tissue injury and risk of death and can cause tissue injury. JCI Insight 2021;6:147610.
161. Montier LL, Deng JJ, Bai Y. Number matters: control of mammalian mitochondrial DNA copy number. J Genet Genomics 2009;36:125-31.
162. Chiu RW, Chan LY, Lam NY, et al. Quantitative analysis of circulating mitochondrial DNA in plasma. Clin Chem 2003;49:719-26.
163. Al Amir Dache Z, Otandault A, Tanos R, et al. Blood contains circulating cell-free respiratory competent mitochondria. FASEB J 2020;34:3616-30.
164. Lindqvist D, Wolkowitz OM, Picard M, et al. Circulating cell-free mitochondrial DNA, but not leukocyte mitochondrial DNA copy number, is elevated in major depressive disorder. Neuropsychopharmacology 2018;43:1557-64.
165. Zhang Q, Raoof M, Chen Y, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010;464:104-7.
166. Kung CT, Hsiao SY, Tsai TC, et al. Plasma nuclear and mitochondrial DNA levels as predictors of outcome in severe sepsis patients in the emergency room. J Transl Med 2012;10:130.
167. Gambardella S, Limanaqi F, Ferese R, et al. ccf-mtDNA as a potential link between the brain and immune system in neuro-immunological disorders. Front Immunol 2019;10:1064.
168. Liu Y, Zhou K, Guo S, et al. NGS-based accurate and efficient detection of circulating cell-free mitochondrial DNA in cancer patients. Mol Ther Nucleic Acids 2021;23:657-66.
169. Ma ML, Zhang H, Jiang P, et al. Topologic analysis of plasma mitochondrial DNA reveals the coexistence of both linear and circular molecules. Clin Chem 2019;65:1161-70.
170. Ma ML, Yakovenko S, Zhang H, et al. Fetal mitochondrial DNA in maternal plasma in surrogate pregnancies: detection and topology. Prenat Diagn 2021;41:368-75.
171. Shibata Y, Kumar P, Layer R, et al. Extrachromosomal microDNAs and chromosomal microdeletions in normal tissues. Science 2012;336:82-6.
172. Turner KM, Deshpande V, Beyter D, et al. Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nature 2017;543:122-5.
173. Kumar P, Dillon LW, Shibata Y, Jazaeri AA, Jones DR, Dutta A. Normal and cancerous tissues release extrachromosomal circular DNA (eccDNA) into the circulation. Mol Cancer Res 2017;15:1197-205.
174. Zhu J, Zhang F, Du M, Zhang P, Fu S, Wang L. Molecular characterization of cell-free eccDNAs in human plasma. Sci Rep 2017;7:10968.
175. Sin STK, Jiang P, Deng J, et al. Identification and characterization of extrachromosomal circular DNA in maternal plasma. Proc Natl Acad Sci USA 2020;117:1658-65.
176. Sin STK, Ji L, Deng J, et al. Characteristics of fetal extrachromosomal circular DNA in maternal plasma: methylation status and clearance. Clin Chem 2021;67:788-96.
177. Sin ST, Deng J, Ji L, et al. Effects of nucleases on cell-free extrachromosomal circular DNA. JCI Insight 2022;7:e156070.
178. Fernández-Domínguez IJ, Manzo-Merino J, Taja-Chayeb L, Dueñas-González A, Pérez-Cárdenas E, Trejo-Becerril C. The role of extracellular DNA (exDNA) in cellular processes. Cancer Biol Ther 2021;22:267-78.
179. Liang H, Peng B, Dong C, et al. Cationic nanoparticle as an inhibitor of cell-free DNA-induced inflammation. Nat Commun 2018;9:4291.
180. Napirei M, Karsunky H, Zevnik B, Stephan H, Mannherz HG, Möröy T. Features of systemic lupus erythematosus in Dnase1-deficient mice. Nat Genet 2000;25:177-81.
181. Martinez-Valle F, Balada E, Ordi-Ros J, Bujan-Rivas S, Sellas-Fernandez A, Vilardell-Tarres M. DNase 1 activity in patients with systemic lupus erythematosus: relationship with epidemiological, clinical, immunological and therapeutical features. Lupus 2009;18:418-23.
182. Thiam HR, Wong SL, Wagner DD, Waterman CM. Cellular mechanisms of NETosis. Annu Rev Cell Dev Biol 2020;36:191-218.
183. Leffler J, Martin M, Gullstrand B, et al. Neutrophil extracellular traps that are not degraded in systemic lupus erythematosus activate complement exacerbating the disease . J Immunol 2012;188:3522-31.
185. Demers M, Wagner DD. Neutrophil extracellular traps: a new link to cancer-associated thrombosis and potential implications for tumor progression. Oncoimmunology 2013;2:e22946.
186. Yang L, Liu Q, Zhang X, et al. DNA of neutrophil extracellular traps promotes cancer metastasis via CCDC25. Nature 2020;583:133-8.
187. Jiménez-Alcázar M, Rangaswamy C, Panda R, et al. Host DNases prevent vascular occlusion by neutrophil extracellular traps. Science 2017;358:1202-6.
188. Zuo Y, Yalavarthi S, Shi H, et al. Neutrophil extracellular traps in COVID-19. JCI Insight 2020;5:138999.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Che H, Stanley K, Jatsenko T, Thienpont B, Vermeesch JR. Expanded knowledge of cell-free DNA biology: potential to broaden the clinical utility. Extracell Vesicles Circ Nucleic Acids 2022;3:216-34. http://dx.doi.org/10.20517/evcna.2022.21
AMA Style
Che H, Stanley K, Jatsenko T, Thienpont B, Vermeesch JR. Expanded knowledge of cell-free DNA biology: potential to broaden the clinical utility. Extracellular Vesicles and Circulating Nucleic Acids. 2022; 3(3): 216-34. http://dx.doi.org/10.20517/evcna.2022.21
Chicago/Turabian Style
Che, Huiwen, Kate Stanley, Tatjana Jatsenko, Bernard Thienpont, Joris Robert Vermeesch. 2022. "Expanded knowledge of cell-free DNA biology: potential to broaden the clinical utility" Extracellular Vesicles and Circulating Nucleic Acids. 3, no.3: 216-34. http://dx.doi.org/10.20517/evcna.2022.21
ACS Style
Che, H.; Stanley K.; Jatsenko T.; Thienpont B.; Vermeesch JR. Expanded knowledge of cell-free DNA biology: potential to broaden the clinical utility. Extracell. Vesicles. Circ. Nucleic. Acids. 2022, 3, 216-34. http://dx.doi.org/10.20517/evcna.2022.21
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 23 clicks
Cite This Article 23 clicks

 Like This Article 18
likes
Like This Article 18
likes







Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.